VI. Chemistry of Sea Water [PDF]
If suspended solid material of either organic or inorganic origin is excluded, sea water may be considered as an aqueous solution containing a variety of dissolved solids and gases. Determination of the chemical nature and concentrations of the dissolved substances is difficult for the following reasons: (1) some of the dissolved substances, such as chloride and sodium ions, are present in very high concentrations, while others, certain metals for instance, are present in such minute quantities that they have not been detected in sea water, although they have been found in marine organisms or salt deposits; (2) two of the major constituents, sodium and potassium, are extremely difficult to determine accurately; (3) it is virtually impossible in some cases to separate related substances such as phosphate and arsenate, calcium and strontium, and chloride, bromide, and iodide. In these cases the combined elements are determined together and usually reported as if they represented only one; that is, calcium and strontium are often calculated as “calcium,” and chloride, bromide, and iodide as “chloride.”
Because of the complex nature of the dissolved materials in sea water a specially developed technique is usually required to determine the concentration of any constituent. The standard methods for the quantitative analysis of solutions which are given in textbooks generally cannot be applied to sea water without adequate checks on their accuracy. This is particularly true when dealing with elements present in extremely low concentrations, because the elements occurring as impurities in the reagents may be in amounts many times those found in the water.
Constancy of Composition
It has been found that, regardless of the absolute concentration of the total solids, the ratios between the more abundant substances are virtually constant. The importance of this result cannot be overemphasized, as upon it depends the validity of the chlorinity:salinity:density relationships and, hence, the accuracy of all conclusions based on the distribution of density where the latter is determined by chemical or indirect physical methods such as electrical conductivity or refractive index.
The relative uniformity in the composition of the sea water was established by the investigations of Forchhammer, Natterer, and Dittmar. Although Forchhammer analyzed a large number of samples, his investigations were not complete because he did not determine certain of the abundant elements. Natterer made more detailed analyses, but it was Dittmar who laid the solid foundation for the present knowledge of the composition of sea water.
Dittmar (1884) made careful determinations on 77 water samples, representative of all oceans, which had been collected on the voyage around the world of H.M.S. Challenger. He determined the halides, sulphate, magnesium, calcium, and potassium. On composite samples he found the ratio of bromine to chlorine and estimated the carbonate. From the sums of the chemical equivalents of the negative and positive ions, he calculated the sodium by difference. This procedure was followed because he was unable to achieve satisfactory direct determinations for sodium. The results of Dittmar's work showed that there were no significant regional differences in the relative composition of sea water; consequently his average values could be used to represent the ratios between the major dissolved constituents. In table 33 are given Dittmar's average values in the units in use at the present time and referred to a chlorinity of 19.00 ‰. The percentages of the various ions are also shown.
| Ion | Original values | Recalculated, 1940 atomic weights | 1940 values | |||
|---|---|---|---|---|---|---|
| Cl = 19 ‰ | % | Cl = 19 ‰ | % | Cl = 19 ‰ | % | |
| C1− | 18.971 | 55.29 | 18.971 | 55.26 | 18.980 | 55.04 |
| Br− | 0.065 | 0.19 | 0.065 | 0.19 | 0.065 | 0.19 |
| SO4− | 2.639 | 7.69 | 2.635 | 7.68 | 2.649 | 7.68 |
| CO3− | 0.071 | 0.21 | 0.071 | 0.21 | …… | …… |
| HCO3− | …… | …… | …… | …… | 0.140 | 0.41 |
| F− | …… | …… | …… | …… | 0.001 | 0.00 |
| H3BO3 | …… | …… | …… | …… | 0.026 | 0.07 |
| Mg++ | 1.278 | 3.72 | 1.292 | 3.76 | 1.272 | 3.69 |
| Ca++ | 0.411 | 1.20 | 0.411 | 1.20 | 0.400 | 1.16 |
| Sr++ | 0.013 | 0.04 | ||||
| K+ | 0.379 | 1.10 | 0.385 | 1.12 | 0.380 | 1.10 |
| Na+ | 10.497 | 30.59 | 10.498 | 30.58 | 10.556 | 30.61 |
| Total | 34.311 | 34.328 | 34.482 | |||
Since 1884 the modification of atomic weights has affected the numerical results reported by Dittmar. Corrections for these changes may be made (Lyman and Fleming, 1940) as shown in the “recalculated” values in table 33. In the latter tabulation the sodium has been recalculated by Difference.
It is interesting to compare Dittmar's results with those obtained by modern methods of analysis as shown in the last columns of the table. The sources of these data are indicated in table 35. It is immediately seen that there are small differences for most of the elements determined by Dittmar and that certain other ions have been added to the list of major constituents. The bound carbon dioxide is reported as bicarbonate ion instead of as carbonate, strontium is given by itself instead of in combination with calcium, and fluoride and boric acid have been added.
The close agreement between the results of Dittmar and those obtained recently is remarkable when we consider the complexity of the problem and the great advance in knowledge of analytical chemistry. However, although the differences are small, they are significant, and hence the importance of Dittmar's work is that it showed the constancy of the ratios between the major constituents, and not that it led to accurate numerical values of these ratios.
In table 33 the composition is shown by referring the substances to a standard concentration, C1 = 19.00 ‰, and by means of the ratios between the different ions and the total dissolved solids. In most instances it is preferable to use a third method; namely, to give the ratios between the various substances and the chlorinity or the chlorosity (p. 52), and these ratios are known as C1-ratios and chlorosity factors, respectively. The Cl-ratio is the amount of any ion or substance per unit (gram) of chlorinity, and is obtained by dividing the concentration in grams per kilogram by the chlorinity, or the concentration in grams per 20°-liter by the chlorosity. Multiplication of the C1-ratio by a given chlorinity or corresponding chlorosity will give the concentrations as grams per kilogram or per liter, respectively. Concentrations in milligram-atom units are always on a liter basis, and, if divided by the chlorosity, yield the ratios that are called chlorosity factors. It may be noted that a chlorosity factor multiplied by chlorinity yields the concentration in milligram-atoms per kilogram.
The uniformity of relative composition in the oceans is the result of circulation and mixing. These operations are continuous, and tend to eliminate regional differences in composition, whatever the cause. Disturbing agencies bring about changes that are small compared to the bulk of the substances present and consequently will not materially affect the relative concentration of the major constituents. Further-more, many of the disturbing processes that tend to modify the relative composition are reversible. For example, the secretion of calcium
The constancy of composition is, as already emphasized, of the greatest importance. Not only is it the basis of the chlorinity:salinity:density relationships, but it also affords a means of estimating the concentrations of all of the major constituents when the concentration of any one of them is known. Furthermore, results of studies on the composition or the physical properties of sea water in any locality are generally applicable to the water in any other part of the oceans.
Except in special areas, such as in the Baltic Sea, the Black Sea, and off the mouths of large rivers, it is not necessary to consider that the water represents special local types with properties that differ from those of sea water in general. Nevertheless, it should be remembered that the composition is not absolutely constant even for the major constituents listed in table 33. Various factors which will be discussed in detail later are always operating and always tend to modify the relative abundances. Rivers introduce dissolved material in proportions that are markedly different from those in the sea, and they also introduce sedimentary material that reacts in various ways with the dissolved constituents. The formation and melting of sea ice may bring about a modified distribution of the dissolved substances.
Thus far, comment has been largely restricted to those constituents of sea water that are present in large, or at least relatively constant, proportions. If we consider those elements which are present in small quantities and which are utilized by marine organisms, the concept of constant composition is no longer generally valid, because the concentrations of these elements vary widely, particularly near the surface. A great part of the work in chemical oceanography is now devoted to determining the space and time variations in variable constituents, and much thought is directed toward the solution of the problems related to the processes that control the observed distribution.
Units Used in Chemical Oceanography
In chemical oceanography most of the numerical results are expressed as concentrations—that is, as the amounts of various constituents in a certain quantity of sea water. Obviously many different combinations
Only two units are to be used for expressing the quantity of sea water: either (1) the kilogram or (2) the amount of water which at 20° C. and pressure one atmosphere occupies the volume of one liter. The latter unit is designated as L20, but in this discussion it will be indicated as L. The system in which the constituents are reported as the amounts present per liter is designated as the “preferred” one, with an alternative for the abundant substances that may be reported as grams per kilogram of sea water. Salinity and chlorinity are always reported as grams per kilogram of sea water. It should be understood that the proposed system applies only to the reporting of analytical data in the literature. Any suitable units may be adopted for the discussion of special problems.
For expressing the amounts of the dissolved constituents, two types of units are proposed: (1) physical units of mass, volume, or pressure, and (2) units based upon the number of atoms of the designated element, which may be present as ions or molecules either singly or in combination with other elements. In certain cases the number of chemical equivalents is acceptable.
The mass units most commonly used are those of the metric system and bear the following relations to each other:
A measure of the number of atoms of the designated element is obtained by dividing the amount of the element, expressed as grams, milligrams, or mygrams, by the gram-atomic weight of the element. Hence, Quantities expressed as gram-, milligram-, or mygram-atoms may be converted to the corresponding mass units by multiplying by the gram-atomic weight of the designated element.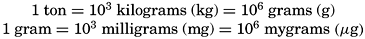
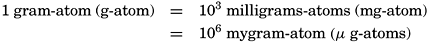
In certain cases (for example, alkalinity and hydrogen-ion concentration) it is desirable to report the concentration in terms of chemical equivalents. The units shall then be
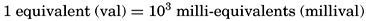
For expressing the partial pressure of gases dissolved in sea water the basic pressure unit is the “physical atmosphere” (p. 55):
Partial pressures shall be expressed in Torr.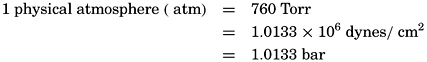
Volume units are all based upon the true liter—that is, the volume of 1 kg of distilled water at 4°C. When volume units are used, the temperature and pressure should be stated. The quantities of dissolved gases, when expressed as milliliters (ml), should be those for 0°C and a pressure of 1 atmosphere, that is, NTP.
The centigrade scale is to be used for reporting temperatures.
The units to be used in reporting data, proposed by the International Association of Physical Oceanography, are given in table 34. It should be noted that all units are based upon the amount of a designated element that may be present either singly (for example, oxygen or calcium) or in combination with other elements (for example, phosphate-phosphorus).
Because the 20° liter is the standard volume unit for expressing the quantity of sea water, glassware should be calibrated for this temperature, and, if practicable, measurements and chemical determinations should be made at or near this temperature. If the sea-water samples are not at 20°, it may be necessary to apply certain corrections. Full descriptions of the methods for making such corrections and tables to facilitate the transformation are included in the Report of the International Association of Physical Oceanography. In most cases the accuracy of the methods of analysis for the elements present in small amounts do not justify such corrections.
As already stated, it is frequently desirable to express the relative concentrations as Cl-ratios or chlorosity factors (p. 167). These relationships may be used to calculate the quantity of the major elements present in water of known chlorinity or to check variations in composition which may be brought about by natural agencies, pollution by sewage and industrial wastes, or by other agencies.
Composition of Sea Water
So far, the discussion of the composition of sea water has been based mainly on the results of the fundamental investigations of Dittmar. Since his time our knowledge of the composition of sea water has increased tremendously. Improved methods of analysis have been developed and consequently more accurate values can be obtained. Tests have also been developed for the detection and determination of elements other than those previously discussed. Particular efforts have been devoted to the study of the so-called plant nutrients—that is, those elements
| Designated substance | Abbreviation | Units (p = preferred, a = alternative) | |||
|---|---|---|---|---|---|
|
|
| ‰ | ||
| Ammonia-nitrogen | Ammonia-N | p | |||
| Argon | Argon | p | |||
| Arsenate-arsenic | Arsenate-As | p | |||
| Arsenite-arsenic | Arsenite-As | p | |||
| Borate-boron | Borate-B | p | |||
| Calcium | Ca | p | a | ||
| Carbon dioxide | Carbon dioxide-C | p | |||
| CO2 | a | ||||
| Chlorinity | Cl | p | |||
| Copper | Cu | p | |||
| Iron | Fe | p | |||
| Magnesium | Mg | p | a | ||
| Manganese | Mn | p | |||
| Nitrate-nitrogen | Nitrate-N | p | |||
| Nitrite-nitrogen | Nitrite-N | p | |||
| Nitrogen (gas) | N2 | p | a | ||
| Oxygen (gas) | O2 | p | a | ||
| Phosphate-phosphorus | Phosphate-P | p | |||
| Potassium | K | p | a | ||
| Radioactive substances | p | ||||
| Salinity | S | p | |||
| Silicate-silicon | Silicate-Si | p | |||
| Sodium | Na | p | a | ||
| Sulphate | Sulphate-S | p | |||
| SO4 | a | ||||
| Hydrogen sulphide | Sulphide-S | p | |||
| H2S | a | ||||
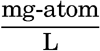 [
[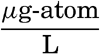 [
[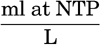 [
[Hence, in order to prepare a tabulation of the composition of sea water it is necessary to combine the results of numerous workers who have examined samples from different sources. All available data were collected by Thompson and Robinson (1932), and additional references will be found in the following discussion. In some cases the information is extensive, but for other elements only a few determinations have been made on water from a single locality. We shall first examine the quantities of the major elements—that is, those which bear a virtually constant relationship to the chlorinity.
In table 35 is given a compilation of the major ions that make up over 99.9 per cent of the known dissolved solid constituents of sea water. The sources of these data have been discussed by Lyman and Fleming (1940). The concentrations of the various ions are shown for water of 19.00 ‰ chlorinity, and also the Cl-ratios. The quantities are also expressed in terms of chemical equivalents per kilogram for water of 19.00 ‰ chlorinity and as milligram-atoms per 20° liter. Chlorosity factors are given for units of milligram-atoms. The carbon dioxide has been reported as bicarbonate. This method is not strictly accurate, because the bound carbon dioxide content of sea water is variable, but, as will be shown in the discussion of the carbon dioxide system, the sum of the chemical equivalents of carbonate and bicarbonate is virtually constant for any chlorinity.
It is immediately seen that the sum of the halides (chloride, bromide, and fluoride) by weight is greater than the chlorinity. The amount of iodide is negligible. Even if the bromide is calculated as chloride, and if the fluoride is disregarded because it does not take part in the chlorinity determination, the chloride equivalent is 1.00045 times greater than the chlorinity. The reasons for this apparent discrepancy have been discussed on page 52.
Lyman and Fleming (1940) obtained the following empirical equation for the dissolved solids as represented in table 35:
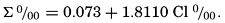
From this it will be seen that in water of 19.00 ‰ chlorinity the total dissolved solids are 34.4816 ‰, but, according to the equation used to calculate the salinity from the chlorinity (p. 51), the salinity is 34.325 ‰. Thus, the total amount of dissolved solids is greater than the salinity. If, on the other hand, the salinity is calculated from the total solids, using the definition for the former quantity—that is, by converting the bicarbonate to oxide and converting the bromide to chloride—we obtain the salinity “by definition” as 34.324 ‰. This agreement must be considered as more or less accidental, as there are many uncertainties in the analytical data. Confidence in the values is strengthened, however, by the fact that the sodium: chlorinity ratio as reported by Robinson and Knapman (1941) agrees exactly with the value that Lyman and Fleming (1940) found by difference. Although the table represents the most probable figures for the composition of the major dissolved constituents, it is subject to change as additional data become available.
| Ion | ‰ | Cl-ratio, g per unit Cl | Equivalent per kg of sea water | mg-atoms per liter | Chlorosity factor, mg-atoms per unit Cl | Authority |
|---|---|---|---|---|---|---|
Total dissolved solids = 34.4816 ‰ Sum of constituents (HCO3− as O−, and Br− as Cl−) = 34.324 ‰ Salinity (S ‰ = 0.030 + 1.805 Cl ‰) = 34.325 ‰ | ||||||
| Chloride, Cl− | 18.9799 | 0.99894 | 0.5353 | 548.30 | 28.173 | Dittmar (1884), Jacobsen and Knudsen (1940) |
| Sulphate, SO4− | 2.6486 | 0.1394 | 0.0551 | (SO4-S) 38.24 | 1.451 | Thompson, Johnston, and Wirth (1931) |
| Bicarbonate, HC03− | 0.1397 | 0.00735[a] | 0.0023 | (HCO3−C) 2.34 | 0.120 | Revelle (1936) |
| Bromide, Br− | 0.0646 | 0.00340 | 0.0008 | 0.83 | 0.0426 | Dittmar (1884) |
| Fluoride, F− | 0.0013 | 0.00007 | 0.0001 | 0.07 | 0.0036 | Thompson and Taylor (1933) |
| Boric acid,c H3BO3 | 0.0260 | 0.00137[b] | [c] | (H3BO3-B)0.43 | 0.0221 | Harding and Moberg (1934), Igelsrud, Thompson, and Zwicker (1938) |
| _______ | ||||||
| Total | 0.5936 | |||||
| Sodium,[d] Na+ | 10.5561 | 0.5556 | 0.4590 | 470.15 | 24.153 | By difference, and Robinson and Knapman (1941) |
| Magnesium, Mg++ | 1.2720 | 0.06695 | 0.1046 | 53.57 | 2.752 | Thompson and Wright (1930) |
| Calcium, Ca++ | 0.4001 | 0.02106 | 0.0200 | 10.24 | 0.5262 | Kirk and Moberg (1933); Thompson and Wright (1930) |
| Potassium, K+ | 0.3800 | 0.02000 | 0.0097 | 9.96 | 0.5113 | Thompson and Robinson (1932) |
| Strontium, Sr++ | 0.0133 | 0.00070 | 0.0003 | 0.15 | 0.0077 | Webb (1938) |
| _______ | ||||||
| Total | 0.5936 | |||||
The data in table 35 apply more specifically to surface water than to deep water. Both bicarbonate ion and calcium will be slightly higher in deeper water. Furthermore, some of the other compounds not included in this tabulation, such as nitrate and silicate, may be present in sufficient quantities to disturb the balance of the anions and cations shown in the table. The Cl-ratios should therefore be considered more as indices than as absolute values. However, in no case will the ratios vary by more than a unit or two in the last decimal place when the water under investigation is from the open sea. Under abnormal conditions, as in highly diluted water, larger departures may be found. By definition the salinity is not zero at zero chlorinity; hence the ratios of certain elements would be expected to approach infinity at very high dilutions when the diluting water contained substances other than halides. Therefore, in studies in areas of highly diluted water the character of the river water should be taken into account. As pollution problems frequently occur in such areas, it will be necessary to determine the normal ratios for different dilutions for a specific zone before any conclusions can be drawn as to the nature or extent of the pollution.
Elements Present in Sea Water
Thus far, only the major constituents of sea water have been considered. In table 36 are entered all elements that are known to occur in sea water as dissolved solids, except hydrogen and oxygen. They are not given as ions in this case but as the amounts of the individual elements which occur in water of chlorinity 19.00 ‰. The elements are arranged in the order of their abundance. In the first column they are reported as milligrams per kilogram, and in the second as milligram-atoms per liter. For convenience, the 1940 atomic weights and their reciprocals have been included. These constants are necessary when converting weight units to gram-atom units, and vice versa. The values for the major elements correspond to those given in previous tables and, in general, are valid for surface water. For many of the elements ranges in concentration have been indicated. No doubt ranges should be shown for others, but the lack of sufficient observations or uncertainty as to the reliability of reported data leaves these problems unsettled. For many of the elements that are present in very low concentrations there are only one or two determinations available, and in some cases only indirect estimates have been made. Hence, in these cases the indicated values can represent only the order of magnitude of the quantities present. Omitting the six most abundant elements, only carbon (CO2 components), silicon,
Forty-four elements are listed in table 36, and if we add hydrogen, oxygen, and the inert gases neon, helium, and argon, we obtain a total of forty-nine elements that are known to occur in sea water. Further investigations will undoubtedly demonstrate the presence of others. Certain problems of the origin and concentration of the dissolved solids relative to their concentration in the earth's crust will be discussed later.
The following brief discussion is limited to those elements that either occur in relatively large amounts or whose distribution has been shown to be affected by biological activity. For elements in the latter group additional data are given in chapter VII. In table 36 references are given for those elements not discussed in the text. A comprehensive discussion is given by Thompson and Robinson (1932), and other results are reported by Goldschmidt (1937) and Wattenberg (1938). The elements are considered in the order in which they appear in the table.
Chlorine, present as chloride ion, is the most abundant ion and makes up about 55 per cent by weight of the dissolved material. It is rarely measured except in combination with other halides in the chlorinity determination. The bromide and iodide are then computed as if they were chloride. It should be kept in mind that the ratio of the chlorine equivalent of the halides to the chlorinity is 1.00045 (p. 52). The chlorinity is of the greatest importance, not only as the basis of density computations, but also as the standard to which those substances present in major amounts are referred.
Sodium is the most abundant cation in sea water, but it is rarely determined directly, owing to the technical difficulties involved in the determination of the alkali metals. The average ratio to chlorinity, 0.5556, as obtained by Robinson and Knapman (1941) agrees exactly with the value that Lyman and Fleming (1940) calculated by difference. It is somewhat higher than the average of 0.5509 given by Thompson and Robinson (1932), but is in fair agreement with the ratio 0.5549 obtained by Webb (1939) by direct analysis. The sodium: chlorinity ratio may be modified near river mouths.
Magnesium content of sea water has been investigated rather carefully, particularly by Thompson and Wright (1930). The magnesium is usually determined by a special modification of the magnesium-ammonium-phosphate method. The ratio of magnesium to chlorinity is very uniform.
| Element | mg/kg Cl = 19.00 ‰ | mg-atoms/L Cl = 19.00 ‰ | Atomic weight (1940) | 1/atomic weight | Authority |
|---|---|---|---|---|---|
| Chlorine | 18980 | 548.30 | 35.457 | 0.02820 | |
| Sodium | 10561 | 470.15 | 22.997 | 0.04348 | |
| Magnesium | 1272 | 53.57 | 24.32 | 0.04112 | |
| Sulphur | 884 | 28.24 | 32.06 | 0.03119 | |
| Calcium | 400 | 10.24 | 40.08 | 0.02495 | |
| Potassium | 380 | 9.96 | 39.096 | 0.02558 | |
| Bromine | 65 | 0.83 | 79.916 | 0.01251 | |
| Carbon | 28 | 2.34 | 12.01 | 0.08326 | |
| Strontium | 13 | 0.15 | 87.63 | 0.01141 | |
| Boron | 4.6 | 0.43 | 10.82 | 0.09242 | |
| Silicon | 0.02 –4.0 | 0.0007 –0.14 | 28.06 | 0.03564 | |
| Fluorine | 1.4 | 0.07 | 19.00 | 0.05263 | |
| Nitrogen (comp.) | 0.01 –0.7 | 0.001 –0.05 | 14.008 | 0.07139 | |
| Aluminum | 0.5 | 0.02 | 26.97 | 0.03708 | |
| Rubidium | 0.2 | 0.002 | 85.48 | 0.01170 | |
| Lithium | 0.1 | 0.014 | 6.940 | 0.14409 | |
| Phosphorus | 0.001–0.10 | 0.00003–0.003 | 30.98 | 0.03228 | |
| Barium | 0.05 | 0.0004 | 137.36 | 0.00728 | |
| Iodine | 0.05 | 0.0004 | 126.92 | 0.00788 | |
| Arsenic | 0.01–0.02 | 0.00015–0.0003 | 74.91 | 0.01335 | |
| Iron | 0.002–0.02 | 0.00003–0.0003 | 55.85 | 0.01791 | |
| Manganese | 0.001–0.01 | 0.00002–0.0002 | 54.93 | 0.01820 | |
| Copper | 0.001–0.01 | 0.00002–0.0002 | 63.57 | 0.01573 | |
| Zinc | 0.005 | 0.00008 | 65.38 | 0.01530 | Atkins (1936) |
| Lead | 0.004 | 0.00002 | 207.21 | 0.00483 | Boury (1938) |
| Selenium | 0.004 | 0.00005 | 78.96 | 0.01266 | Goldschmidt and Strock (1935) |
― 177 ― Cesium | 0.002 | 0.00002 | 132.91 | 0.00752 | Wattenberg (1938) |
| Uranium | 0.0015 | 0.00001 | 238.07 | 0.00420 | Föyn et al (1939) |
| Molybdenum | 0.0005 | 0.000005 | 95.95 | 0.01042 | Ernst and Hoermann (1936) |
| Thorium | <0.0005 | <0.000002 | 232.12 | 0.00431 | Föyn et al (1939) |
| Cerium | 0.0004 | 0.000003 | 140.13 | 0.00714 | Goldschmidt (1937) |
| Silver | 0.0003 | 0.000003 | 107.880 | 0.00927 | Haber (1928) |
| Vanadium | 0.0003 | 0.000006 | 50.95 | 0.01963 | Ernst and Hoermann (1936) |
| Lanthanum | 0.0003 | 0.000002 | 138.92 | 0.00720 | Goldschmidt (1937) |
| Yttrium | 0.0003 | 0.000003 | 88.92 | 0.01125 | Goldschmidt (1937) |
| Nickel | 0.0001 | 0.000002 | 58.69 | 0.01704 | Ernst and Hoermann (1936) |
| Scandium | 0.00004 | 0.0000009 | 45.10 | 0.02217 | Goldschmidt (1937) |
| Mercury | 0.00003 | 0.0000001 | 200.61 | 0.00498 | Goldschmidt (1937) |
| Gold | 0.000006 | 0.00000002 | 197.2 | 0.00507 | Haber (1928) |
| Radium | 0.2 – 3 × 10−10 | 0.8 – 12 × 10−13 | 226.05 | 0.00442 | Evans, Kip, and Moberg (1938) |
| Cadmium | Fox and Ramage (1931) | ||||
| Chromium | Webb (1937) | ||||
| Cobalt | Thompson and Robinson (1932) | ||||
| Tin | Thompson and Robinson (1932) |
Sulphur is present in sea water as sulphate ion, and is in this form usually determined by precipitation as barium sulphate. An extensive study of the sulphate distribution has been made by Thompson, Johnston, and Wirth (1931). Under stagnant conditions occurring in certain isolated basins, and in and near bottom sediments, a part of the sulphate may be converted to sulphide ion. Considerable quantities of sulphide occur in the Black Sea and in certain Norwegian fjords, and its presence has been reported in many localities. The sulphate: chlorinity ratio may also be modified by dilution with river water, which is generally relatively high in sulphate. Processes of freezing and melting may possibly affect the relative concentration (p. 216).
Calcium is present in much smaller quantities than either sodium or magnesium, but its distribution in the ocean has been studied much more thoroughly, mainly because calcium is a major constituent of many skeletal remains found in marine sediments. By deposition of such remains calcium is permanently removed from the water, but this removal does not necessarily imply that the calcium concentration is decreasing, because a large supply is maintained by the river waters flowing into the sea. Detectable differences in the calcium: chlorinity ratio have been observed. In the Baltic, Gripenberg (1937a) has shown that the type of river water which has diluted the sea water can be determined from that ratio. Furthermore, Moberg and Revelle (1937) have demonstrated the existence of vertical differences in the calcium: chlorinity ratio which they attribute to the removal of calcium in the surface layers through biological activity. Interest in the concentration of calcium has also centered around the question of the solubility of calcium carbonate in sea water and the factors that control precipitation and solution. In certain areas calcium carbonate is apparently precipitated inorganically, and in other regions it apparently passes into solution. In addition to these problems, knowledge of the calcium concentration is important in an understanding of the carbon dioxide system in the sea, which will be discussed later. The quantity of calcium is usually determined by precipitation as the oxalate under carefully controlled conditions and subsequent titration with potassium permanganate. One such method has been described by Kirk and Moberg (1933).
Webb has pointed out that in this method for the estimation of calcium the strontium will be carried down, and hence the calcium figure will be too high by the equivalent amount of strontium. As the ratio calcium: strontium is apparently constant, Webb suggests that the “calcium” shall be taken to mean the calcium after the strontium and barium have been replaced by calcium. Since the barium is negligible in this case, the values of “calcium” will be given directly by volumetric methods, but when the quantities are determined by weighing, corrections
Potassium is the fourth most abundant cation and is present in amounts of only a few per cent of that of sodium. The potassium is rarely determined directly, but apparently it bears a very constant relationship to the chlorinity (Thompson and Robinson, 1932). However, the content of potassium may be modified by biological agencies, since some organisms, particularly the large algae, concentrate potassium to a marked degree. The ratio of the potassium to chlorinity may also be modified by dilution with river water. The potassium may react with the colloidal and clay particles brought to the sea by rivers and run-off, and consequently this agency may influence the ratio. Certain minerals formed on the sea bottom, such as glauconite, contain potassium.
Bromine shows a very constant ratio to the chlorinity and is apparently all present as bromide ion.
Discussion of the concentration of carbon in sea water is complicated by the fact that it occurs not only in the form of carbonic acid and its salts but also in appreciable amounts as a constituent of organic material, either living or dead. The detrital organic material may be either particulate or in solution. The solubility of carbon dioxide depends upon the temperature and salinity of the water, and exchange of carbon dioxide with the atmosphere takes place at the surface. Photosynthesis in the surface layers reduces the amount of carbon dioxide in the water, and respiration increases the concentration. Consequently, the quantities of carbon present as either free carbon dioxide, bicarbonate, or carbonate will show a considerable range. These problems will be discussed in the sections dealing with the carbon dioxide system in the sea. The quantity of carbon given in table 36 was calculated on the assumption that only bicarbonate ions were present. The organic carbon, which is probably of the order of 2 to 3 mg/L (0.15 to 0.25 mg-atoms/L), was not included. The methods by which the different carbon dioxide components and the particulate and dissolved organic carbon may be determined are discussed later.
Strontium has not been investigated in detail, as it is extremely difficult to determine quantitatively. In determinations of calcium by means of the oxalate precipitation, the strontium is carried down with the calcium, and consequently the ratio of calcium: chlorinity usually reported for sea water represents the calcium plus strontium reported as calcium. Strontium is a constituent of the calcareous skeletons of certain organisms.
Boron occurs in sea water in a surprisingly high concentration and bears a constant relationship to the chlorinity. Apparently it is present as undissociated boric acid. There has been considerable uncertainty as to the form in which boron occurs, but the method of determination is standardized against boric acid and the values can at least be expressed as equivalent to a certain concentration of boric acid. The determination of boric acid in sea water is based on titration with very dilute sodium hydroxide in the presence of mannitol. Methods have been described by Harding and Moberg (1934) and by Igelsrud, Thompson, and Zwicker (1938). The amount of boron present in sea water is of interest in the carbonate equilibria and in this connection will be discussed later. Boron is concentrated by certain marine organisms.
Silicon has been studied extensively because it is utilized by diatoms and other silica-secreting organisms. According to a tabulation by Thompson and Robinson (1932), the silicate-silicon varies by more than one hundredfold—namely, from 0.0007 to 0.11 mg-atoms/L (0.02 to 3.0 mg/L). Clowes (1938) found values slightly exceeding .14 mg-atoms/L (4.0 mg/L) in the deep waters of the Antarctic. Surface samples are usually low, owing to the development of silica-secreting organisms, but a progressive increase in silicate takes place with depth, which is ascribed to the dissolving of soluble silicates. However, there is always the possibility that the water contains silicon in some compound present in colloidal form. River water contains a high content of silicon, both in solution and as colloidal particles. Diatom and radiolarian oozes contain the siliceous remains of organisms that have developed near the surface and settled to the bottom after their death. Although siliceous deposits of organic origin cover large areas, most of the siliceous skeletal remains dissolve after the death of the organisms. Silicon present as soluble silicate is determined colorimetrically. The method has been described by Thompson and Houlton (1933) and by Wattenberg (1937). Because of the rapidity with which water samples are contaminated by silicate that dissolves from the glass, the analyses should be made soon after the water samples are collected. Waxed containers are sometimes recommended, and it is always desirable to use “aged” bottles that have been thoroughly leached with sea water. Tourky and Bangham (1936) tested the reaction between the molybdate reagent and colloidal silica and found that the color development was not proportional to the amount of silicon present. Treatment of the colloidal silica with alkali prior to analysis yielded correct values. Experiments with sea water indicated that colloidal silica may pass into true solution on ageing.
Fluorine is present in oceanic sea water in concentrations slightly above 1 mg/L. It is present as fluoride and, according to the work of Thompson and Taylor (1933), bears a constant ratio to the chlorinity.
The method of determination is described by these authors. Little is known concerning the role of fluorine in the sea.
Nitrogen occurs in sea water both in compounds of various kinds and as free dissolved nitrogen gas. As it is an essential constituent of living matter, nitrogen is found in organic compounds both in organisms and in particulate and dissolved organic material in amounts between 0.1 and 10.0 μg-atoms/L (p. 254). In addition, it is present as nitrate, nitrite, and ammonia. In routine observations only the inorganic nitrogen compounds are determined. Nitrate- and nitrite-nitrogen are determined colorimetrically, and the ammonia either colorimetrically (Robinson and Wirth, 1934) or by micro-titration after distillation (Krogh, 1934).
The nitrate method originally described by Harvey (1926) is given by Wattenberg (1937). Rakestraw (1936) and Wattenberg describe the procedure for the determination of nitrite. Since the inorganic nitrogen compounds are subject to change after the water samples have been collected, analyses must be run within a few hours. Even the addition of preservatives may not prevent changes in the NH3 and NO2, indicating that purely chemical transformations may be involved. Ammonia tends to disappear in storage, and nitrite sometimes decreases, but at other times shows an increase. The nitrate, which is more abundant, does not show such relatively large changes.
Because of their relatively low concentrations and their utilization by organisms, the inorganic nitrogen compounds show a wide range in values:
The distribution of nitrate in the oceans has been and is studied a great deal, as it may limit the production of phytoplankton when it is reduced to minimal quantities in the surface layers. Nitrate-nitrogen usually shows a subsurface maximum at a depth of several hundred meters. Nitrite nitrogen has a peculiar distribution and is generally found in a rather thin stratum in or above the thermocline. Lessi is known concerning the distribution of ammonia, as it is not so readly measured as the other inorganic compounds of nitrogen, but it is apparently rather uniform throughout the water column.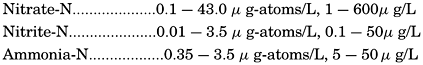
Nitrogen compounds are carried to the sea by rivers and by precipitation. The greater part of these are supposed to have been fixed by electrical discharges in the atmosphere. Possibly a certain amount of the fixed nitrogen in the sea is liberated as free nitrogen and returned to the atmosphere. Bottom sediments contain a small percentage of organic nitrogen in resistant organic detritus, and a part of this is
Aluminum is present in sea water in very small amounts. The colloidal clay particles which are carried to the sea contain a large percentage of aluminum, and hence analyses of water samples collected near shore may show the presence of aluminum, but it is not necessarily all in solution. The value given in table 36 is the average quantity reported by Haendler and Thompson (1939). Their values range between 0.006 and 0.065 mg-atoms/L (0.16 and 1.8 mg/L) with an average of 0.02 mg-atoms/L (0.54 mg/L).
Although earlier workers (Thompson and Robinson, 1932) were unable to detect rubidium in sea water, Goldschmidt (1937) has reported about 0.002 mg-atoms/L (0.2 mg/L).
Lithium content of sea water has been investigated by Thomas and Thompson (1933), who found 0.014 mg-atoms/L (0.1 mg/L).
Phosphorus, which is present in sea water as phosphate ions, is another of the essential constituents of living organisms, and its distribution in the sea is markedly affected by organic agencies. In addition to the nitrogen and silicon compounds, phosphate-phosphorus has been considered as one of the substances that may limit production of plant life. The inorganic phosphorus concentration varies from virtually zero at the surface, under certain conditions, to approximately 0.003 mg-atoms/L (0.090 mg/L) at subsurface levels when values are corrected for salt error. There is frequently a subsurface maximum similar to that in the distribution of nitrate-nitrogen. Phosphorus removed from the surface layers by phytoplankton is largely returned to solution on the death and decomposition of the organisms. It is supplied by rivers, and some is removed from the sea, as a small quantity is present in most marine sediments. In certain shallow areas, phosphatic concretions are found that contains a rather high concentration of phosphorus. The mode of origin of these concretions is not yet known. It has been suggested that in many regions the water is supersaturated in respect to tricalcium phosphate which, therefore, may be deposited inorganically (Dietz, Emery, and Shepard, 1942).
Phosphate phosphorous is determined colorimetrically. The method has been described by Robinson and Wirth (1935) and Wattenberg (1937). Cooper (1938a) has discussed the magnitude of the salt error. Phosphate analyses are frequently carried out as routine observations, and our knowledge of the distribution of phosphate in the ocean is fairly comprehensive. The rather scant knowledge we have concerning the
The amount of barium in sea water has been reported by Goldschmidt (1937) as 0.0004 mg-atoms/L (0.05 mg/L). This is lower than the values reported by Thompson and Robinson (1932). Barium occurs in marine organisms and it is a constituent of most marine sediments. In certain localities the deposits contain large amounts of barium sulphate in the form of concretions and nodules. The mode of formation of these structures is not yet understood.
The distribution and concentration of iodine in the sea has received a great deal of attention because of its important role in the physiology of man and terrestrial animals. Marine products are an important source of iodine-rich foods, The form in which iodine occurs in sea water is not yet clearly understood, but at least part of it is present as iodide and iodate. It is concentrated to a marked degree by marine plants, and for many years sea weeds have been used as a commercial source of iodine. The distribution and determination of iodine in sea water and marine organisms have been discussed by Closs (1931) and Reith (1930).
Arsenic content of sea water has been investigated by Rakestraw and Lutz (1933), who report values ranging from 0.15 to 0.3 μg-atoms/L (9 to 22 μg/L). This wide range is attributed to the fact that organisms may utilize arsenic in place of phosphorus. It is known to be a constituent of the tissues of many marine forms. The exact form in which arsenic occurs in sea water is not yet known.
Iron is an essential constituent of plants and has been considered as one of the substances that may limit the amount of plant production in the sea. Investigations show that at least part of the iron is not present in true solution, as it can be removed by ultrafiltration. Cooper (1937b) has pointed out that the amount of iron in true solution as ferric or ferrous salts is probably less than 2 μg/L, whereas the total iron present is generally about ten times this quantity. The amount present in the plankton may be as much as 16 per cent of the total iron of the water. Harvey (1937) considers that diatoms are able to adsorb and utilize colloidal iron. Iron is brought to the sea in relatively large quantities in the colloidal clay particles, and consequently considerable amounts of iron are found in the marine sediments. In many instances the iron content of the sediments is even higher than should be expected, indicating addition of iron through physical, chemical, or organic agencies. In inshore areas near the source of supply the total iron content of the water is sometimes much higher than that found in the open ocean. Methods for the determination of iron in sea water in its various forms have been described by Thompson and Bremner (1935a and b), Cooper (1935), and Rakestraw, Mahnke, and Beach (1936).
Manganese is apparently subject to concentration by marine organisms. Thompson and Wilson (1935) have reported values between 0.02 and 0.2 mg-atoms/L (1 and 10 mg/L). The value cited by Goldschmidt (1937) is 4 mg/L. Interest in manganese has been aroused by the occurrence of manganese nodules which are widely distributed in certain types of marine sediments, particularly in the Pacific Ocean.
The quantity of copper present in sea water probably lies between 0.02 and 0.2 μg-atoms/L (1 to 10 μg/L) (Marks, 1938, Wattenberg, 1938). Copper is an essential constituent of many marine organisms and it is also considered a factor in the life history of oysters, as a relatively high copper content of the water is apparently necessary for proper development of the larvae.
Much interest is attached to the content of radioactive elements in sea water, because deep-sea sediments are high in radium, compared to igneous rocks, and it is considered that the enrichment must be due to precipitation from the water of radium or its precursors. The radium content of sea water has been studied by many investigators, using various techniques, but it is only recently that methods have been sufficiently refined to yield trustworthy results. Studies by Evans, Kip, and Moberg (1938) and by Pettersson and Rona (Föyn et al, 1939) show that the radium content, measured by the radon emanation technique, varies between about 0.2 and 3.0 × 10−13 ‰ in sea water of salinity approximately 35 ‰. The low values are found in the surface layers, and it is suggested that organisms are responsible for a selective removal of this element. Both groups of workers found that organisms concentrate the radium about one hundredfold in their soft tissues. Calcareous structures show an increase in the radium:calcium ratio over that in the water. The maximum value listed above—namely 3.0 × 10−13 ‰— was found in water in contact with the sediments (Evans, Kip, and Moberg, 1938), and generally the radium content of the deeper waters is about 1 × 10−13 ‰.
Pettersson and co-workers (Föyn et al, 1939) have emphasized the importance of searching for the radioactive precursors of radium, as this element has the relatively short half-life period of only 1690 years. Of these elements uranium and ionium are probably the most; important, but thus far only uranium has been examined. Karlik (Föyn et al 1939) has analyzed a number of samples from various parts of the oceans and obtained for oceanic water a mean value of 1.5 × 10−6 ‰. Surface waters have a somewhat lower content than those from greater depths, but Karlik does not consider that the data are sufficiently adequate to show any differential removal. Studies of the dilute waters of the Baltic Sea showed that the uranium content was a function of the salinity.
Föyn and Rona (Föyn et al) have sought for thorium in sea water, but have been unable to detect it by the most refined methods. By examining
The radium content of marine sediments and the theories concerning the deposition of radium and its precursors are discussed in the chapter on marine sedimentation.
Preparation of Artificial Sea Water
It is impossible to prepare solutions that exactly duplicate the properties of sea water because (1) the ions (salts) in which the elements occur in sea water are not always known, (2) elements that occur in sea water in small amounts are present as contaminants in other compounds in quantities which may far exceed those that should be added, and (3) many of the salts which must be added in fairly large amounts are hygroscopic or contain water of crystallization and are difficult to weigh accurately. The latter difficulty may be partially avoided by preparing concentrated solutions of these salts, determining their concentration by chemical analysis, and adding the required volume of the solution.
Although it would be of great interest to prepare solutions duplicating all the physical and chemical properties of sea water, it is generally not essential. In studies of certain of the physical-chemical properties, it is sufficient to add to the solution only the more abundant ions. In other instances—for example, when chemical methods are to be standardized—only one element or ion need be accurately known and other ions only approximately. Furthermore, in experiments with marine plants the major elements may not have to be closely controlled, but it will generally be necessary to know the concentrations of the biologically essential elements that are normally present in small amounts. If possible, natural sea water should always be used in physical or biological studies, but in the latter case it is sometimes desirable to enrich the water with certain of the plant nutrients (p. 235). Rogers (1938) has discussed various “modified ” types of solutions that are used in experiments on marine animals.
In table 37 are given three suggested formulae for preparing solutions approximating the composition of sea water. They have been adjusted to yield solutions of 19.00 ‰ chlorinity. The recipe of McClendon et al (1917), which has been used quite extensively, contains the nitrogen, phosphorus, and silicon needed by marine plants. Additional elements may be necessary but are probably always present as impurities. The formulae of Brujewicz (Subow, 1931) and of Lyman and FIeming (1940) contain only the major elements. The last-mentioned recipe corresponds to the composition of sea water given in table 35. The other formulae have not been adjusted to the composition presented in earlier sections of
| McClendon et al (1917) | Brujewicz (Subow, 1931) | Lyman and Fleming (1940) | |||
|---|---|---|---|---|---|
| Salt | g/kg | Salt | g/kg | Salt | g/kg |
| NaCl | 26.726 | NaCl | 26.518 | NaCl | 23.476 |
| MgCl2 | 2.260 | MgCl2 | 2.447 | MgCl2 | 4.981 |
| MgSO4 | 3.248 | MgSO4 | 3.305 | Na2SO4 | 3.917 |
| CaCl2 | 1.153 | CaCl2 | 1.141 | CaCl2 | 1.102 |
| KCl | 0.721 | KCl | 0.725 | KCl | 0.664 |
| NaHCO3 | 0.198 | NaHCO3 | 0.202 | NaHCO3 | 0.192 |
| NaBr | 0.058 | NaBr | 0.083 | KBr | 0.096 |
| H3BO3 | 0.058 | H3BO3 | 0.026 | ||
| Na2SiO3 | 0.0024 | SrCl2 | 0.024 | ||
| Na2Si4O9 | 0.0015 | NaF | 0.003 | ||
| H3PO4 | 0.0002 | ||||
| Al2Cl6 | 0.013 | ||||
| NH3 | 0.002 | ||||
| LiNO3 | 0.0013 | ||||
| Total | 34.4406 | 34.421 | 34.481 | ||
| Water to 1,000.0000 | Water to 1,000.000 | Water to 1,000.000 | |||
Dissolved Gases in Sea Water
All of the atmospheric gases are found in solution in sea water. In addition to nitrogen and oxygen, the most abundant gases in the air, carbon dioxide is present in large quantities in sea water, chiefly combined as carbonates and bicarbonates. Of the rarer gases, ammonia, argon, helium, and neon have been reported in sea water, and hydrogen is undoubtedly present in minute quantities. In the absence of dissolved oxygen, hydrogen sulphide may be present, and it is possible that in stagnating water other products of putrefactive decomposition, such as methane, may occur.
Because of its importance in biological processes the dissolved oxygen distribution in the oceans has been examined intensively. Besides being an index to the biological history of the water, the general character of the distribution of oxygen in the deeper water is helpful in studies of currents and of mixing processes. The carbon dioxide distribution is of equal biological importance; its discussion begins on p. 192. Nitrogen has not been studied very widely, as it is apparently chemically inert. Argon is also inert, and is sometimes included with the nitrogen when the
Determination of Dissolved Gases. The content of dissolved oxygen is usually determined by the Winkler method, which depends upon the oxidation of manganous hydroxide by the dissolved oxygen. When acid is added, the oxidized manganese reacts with potassium iodide and sets free iodine, in amounts equivalent to the original dissolved oxygen content, which is determined by titration with sodium thiosulphate. The Winkler method is simple and extremely accurate if certain precautions are observed in handling the water samples and reagents (Thompson and Robinson, 1939).
Problems relating to the determination of carbon dioxide are discussed on p. 192.
Dissolved nitrogen cannot be determined by direct chemical methods, and hence gasometric techniques must be used. In general, the sea-water sample is acidified and all the gases are driven off by boiling or by applying a vacuum. The carbon dioxide is then absorbed in alkali, and the oxygen is absorbed in alkaline pyrogallol. The residual gas is sometimes considered as “atmospheric nitrogen,” although actually there are other gases, principally argon, mixed with it. Rakestraw and Emmel (1937) developed a method for determining the dissolved oxygen and nitrogen content of sea water by first extracting the gases and removing the carbon dioxide, then absorbing the oxygen on phosphorus and the nitrogen on molten lithium. The oxygen contents determined in this way agreed with direct Winkler analyses. The nitrogen determinations on saturated water samples showed results consistently lower than the saturation values according to Fox (1907); further studies (Rakestraw and Emmel, 1938b) indicate that Fox's tables are slightly in error. The gases remaining after the extraction of nitrogen are considered as “argon.”
The presence of hydrogen sulphide can be detected by its characteristic odor. A method for its determination has been described by Gaarder (1916). Although commonly referred to as hydrogen sulphide, a part, at least, will not be present as free gas but as sulphide or bisulphide of some base. A hydrogen sulphide system somewhat comparable to the carbon dioxide system must exist, but it has not yet been investigated.
The determination of ammonia is discussed in the section dealing with nitrogen compounds.
The units to be used in reporting the concentrations of dissolved gases are mg-atoms/L or (ml of gas at NTP)/L.
In some cases it is of interest to know the excess or deficiency of the concentration with respect to water of the same temperature and salinity in equilibrium with the normal dry atmosphere. The saturation values for oxygen and nitrogen are given in tables 38 and 39. If the saturation
| Chlorinity (‰) | 15 | 16 | 17 | 18 | 19 | 20 |
|---|---|---|---|---|---|---|
| Salinity (‰) | 27.11 | 28.91 | 30.72 | 32.52 | 34.33 | 36.11 |
| Temperature (°C) | ||||||
| –2 | 9.01 | 8.89 | 8.76 | 8.64 | 8.52 | 8.39 |
| 0 | 8.55 | 8.43 | 8.32 | 8.20 | 8.08 | 7.97 |
| 5 | 7.56 | 7.46 | 7.36 | 7.26 | 7.16 | 7.07 |
| 10 | 6.77 | 6.69 | 6.60 | 6.52 | 6.44 | 6.35 |
| 15 | 6.14 | 6.07 | 6.00 | 5.93 | 5.86 | 5.79 |
| 20 | 5.63 | 5.56 | 5.50 | 5.44 | 5.38 | 5.31 |
| 25 | 5.17 | 5.12 | 5.06 | 5.00 | 4.95 | 4.86 |
| 30 | 4.74 | 4.68 | 4.63 | 4.58 | 4.52 | 4.46 |
| * mg-atoms of oxygen per liter = 0.08931 × ml/L. | ||||||
| Chlorinity (‰) | 15 | 16 | 17 | 18 | 19 | 20 | 21 |
|---|---|---|---|---|---|---|---|
| Salinity (‰) | 27.11 | 28.91 | 30.72 | 32.52 | 34.33 | 36.11 | 37.94 |
| Temperature (°C) | |||||||
| 0 | 15.22 | 15.02 | 14.82 | 14.61 | 14.40 | 14.21 | 14.01 |
| 5 | 13.43 | 13.26 | 13.10 | 12.94 | 12.78 | 12.62 | 12.45 |
| 10 | 12.15 | 12.00 | 11.86 | 11.71 | 11.56 | 11.42 | 11.27 |
| 15 | 11.04 | 10.92 | 10.79 | 10.66 | 10.53 | 10.39 | 10.26 |
| 20 | 10.08 | 9.98 | 9.87 | 9.76 | 9.65 | 9.54 | 9.43 |
| 25 | 9.30 | 9.21 | 9.11 | 9.02 | 8.92 | 8.82 | 8.73 |
| 28 | 8.89 | 8.84 | 8.72 | 8.62 | 8.53 | 8.44 | 8.35 |
| * mg-atoms of nitrogen per liter = 0.08929 × ml/L. | |||||||
The dissolved oxygen in the sea varies between zero and 0.75 mg-atoms/L (about 8.5 ml/L), although in areas of low temperature and intense photosynthesis the content may exceed this upper limit. Nitrogen, which is apparently unaffected by biological processes, varies between 0.75 and 1.3 mg-atoms/L (8.4 and 14.5 ml/L). The total
Factors Controlling the Distribution of Dissolved Gases. The following general factors control the distribution of dissolved gases in the oceans: (1) temperature and salinity, which determine the concentrations when the water is at the surface and in equilibrium with the atmosphere, (2) biological activity, which markedly affects the concentrations of oxygen and carbon dioxide, (3) currents and mixing processes, which tend to modify the effects of biological activity through mass movement and eddy diffusion.
Water in contact with the atmosphere will tend to reach equilibrium either by giving up or absorbing the individual gases until the water is just saturated. Although the zone of contact is a thin one, convective movements due to cooling, evaporation, or wind action may bring a layer of considerable thickness into equilibrium with the atmosphere. According to Henry's law the concentration, m, of a gas in a liquid is related to the partial pressure, p, of the gas and to the character of the gas and the liquid: m = csp. The numerical value of cs, the coefficient of saturation (absorption), depends upon the units for expressing the concentration of the gas in the solution and its pressure, and upon the chemical character of the gas and the temperature and salinity of the water.
| Gas | Percent of volume or pressure | Partial pressure, Torr |
|---|---|---|
| Nitrogen | 78.03 | 593.02 |
| Oxygen | 20.99 | 159.52 |
| Argon | 0.94 | 7.144 |
| Carbon | 0.03 | 0.228 |
| Hydrogen, | 0.01 | 0.088 |
| 100.00 | 760.000 |
With the exception of water vapor the relative composition of the atmosphere can be considered for practical purposes as constant (table 40). This does not strictly apply to carbon dioxide, relatively slight changes in the partial pressure of which have a pronounced effect upon
The solubilities of those gases, such as oxygen and nitrogen, which do not react chemically with the water or its dissolved salts decrease with increasing temperature and salinity. The solubilities of oxygen and nitrogen in sea water of different salinities over the normal range of temperature were investigated by Fox (1907, 1909). Fox's values for oxygen are still the accepted standards, but his data for nitrogen have been superseded by those of Rakestraw and Emmel (1938b). The solubility of carbon dioxide is greater than that of oxygen and nitrogen because it reacts with the water. Part of the carbon dioxide is present as free CO2 and H2CO3, but in sea water by far the greater part is present as carbonates and bicarbonate, and for the same partial pressure the total CO2 content of sea water is much greater than that of distilled water or neutral salt solutions. The content of free CO2 and H2CO3 decreases with increasing temperature and salinity. Argon is sometimes included with the “atmospheric nitrogen,” and, because its solubility differs from that of nitrogen, the values of the saturation coefficients will be slightly modified. Little is known concerning the other gases in sea water; however, both hydrogen sulphide and ammonia are very soluble gases and their saturation values can play no important part in their distribution.
In table 41 are given values of the saturation coefficients (absorption coefficients) for oxygen, nitrogen, and carbon dioxide in fresh and sea water at different temperatures. The values for oxygen are from Fox (1909), as are also the values for nitrogen in distilled water. The other nitrogen values are from Rakestraw and Emmel(1938b). The values for carbon dioxide (Buch et al, 1932) correspond to the total CO2 in water of zero alkalinity or to the free CO2 and H2CO3 in sea water. It is seen that carbon dioxide is much more soluble than the other two gases and that oxygen is about twice as soluble as nitrogen.
From table 41 it is seen that within the range of chlorinity normally encountered in the oceans the temperature is the most important property influencing the solubility (see also tables 38, 39).
In studies of the distribution of dissolved gases in the sea it is generally assumed that, whatever the location of a water particle, at some time it has been at the surface and in equilibrium with the air. In their studies of the dissolved nitrogen content Rakestraw and Emmel (1938a) have found that the water is virtually saturated (referred to a normal atmosphere), regardless of depth; therefore this assumption appears valid and also indicates that biological activity involving either fixation or production of nitrogen cannot be sufficient to affect significantly the concentration of this gas in the water. As the waters of the oceans appear to have been saturated with oxygen and carbon dioxide at some stage in their history when they were at the surface, the differences between the saturation values (computed from the temperatures and salinities) and the observed contents are measures of the changes which have been effected by biological agencies. The factors influencing the distribution of carbon dioxide are discussed in the following sections, and the distribution of dissolved oxygen will be considered in many places in the ensuing chapters.
| Temperature | 0° | 12° | 24° | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chlorinity (‰) | O2 | N2 | CO2 | O2 | N2 | CO2 | O2 | N2 | CO2 | |||||||||
| ml/L | mg-atoms O/L | ml/L | mg-atoms N/L | ml/L | mg-atoms C/L | ml/L | mg-atoms O/L | ml/L | mg-atoms N/L | ml/L | mg-atoms C/L | ml/L | mg-atoms O/L | ml/L | mg-atoms N/L | ml/L | mg-atoms C/L | |
| 0 | 49.24 | 4.40 | 23.00 | 2.06 | 1715 | 77.0 | 36.75 | 3.28 | 17.80 | 1.59 | 1118 | 50.2 | 29.38 | 2.62 | 14.63 | 1.31 | 782 | 35.1 |
| 16 | 40.1 | 3.60 | 15.02 | 1.73 | 1489 | 66.8 | 30.6 | 2.75 | 11.56 | 1.33 | 980 | 44.0 | 24.8 | 2.22 | 9.36 | 1.08 | 695 | 31.2 |
| 20 | 38.0 | 3.40 | 14.21 | 1.64 | 1438 | 64.5 | 29.1 | 2.61 | 10.99 | 1.26 | 947 | 42.5 | 23.6 | 2.12 | 8.96 | 1.03 | 677 | 30.4 |
The Carbon Dioxide System
Although an extensive literature exists concerning the carbon dioxide system in sea water, publications prior to about 1929 are now chiefly of historic interest. The solution of the problems involved awaited not only the development of suitable analytical methods for the determination of the total carbon dioxide and the various forms in which it is present in sea water, but also the development of the theory and methods for studying the hydrogen ion concentration and certain general theories in physical chemistry. In the brief discussion to follow, only the salient features of the contemporary theories will be presented. These may be adequate for many purposes, but the investigations are not yet closed. Methods of analysis require further refinements, and in many cases fundamental constants must be more accurately determined.
Early investigators studying the carbon dioxide in sea water attempted to apply methods similar to those used for fresh water, where the carbon dioxide is largely present as free carbon dioxide that can be driven off by boiling, by applying a vacuum, or by bubbling through the water a stream of CO2-free gas. The use of such methods on sea water gave variable and conflicting results. It was later found that in order to drive off all the CO2 a strong acid must be added to the water, indicating that at least part of the carbon dioxide was present as the carbonate or bicarbonate of some basic cation. Methods were then developed for the determination of the total carbon dioxide and also for measuring the quantity present as carbonate and bicarbonate ions. It is now considered that the CO2 can exist in the following forms in sea water and that under any given set of conditions equilibria will prevail:
If the gases in sea water are driven off by some suitable method, the CO2 present as dissolved gas will be removed and the equilibria will be displaced until virtually all of the free CO2 and carbonic acid are removed and the bicarbonate is all converted to carbonate. If a strong acid is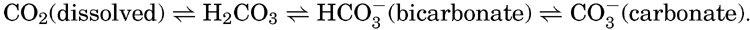
From the foregoing discussion it can be seen that the total CO2 in sea water does not follow Henry's law for the solution of gases in inert solutions. Nevertheless, the partial pressure of the carbon dioxide in sea water in contact with the atmosphere will tend to establish equilibrium with that in the air. If the pressure is increased the amount in solution will be greater, and if it is reduced the quantity of CO2 will decrease. The quantity present in a sample of water in equilibrium with a given carbon dioxide pressure will depend on the concentration of carbon dioxide bound base and the temperature and salinity of the water sample. If these factors are kept constant, the partial pressure of CO2 can be used as a measure of the total carbon dioxide content of the water.
Hydrogen Ion Concentration (pH) of Sea Water. Sea water is normally alkaline. Since both the H+ and OH− ions play parts in the equilibria, any understanding of the carbon dioxide system requires knowledge of their concentrations. Pure distilled water dissociates into hydrogen and hydroxyl ions:
The ionic product [H+] × [OH−], when the concentrations are expressed in chemical equivalents per liter, varies somewhat with temperature, but at 25°C is 10−14 (p. 198). In pure water or in any solution that contains equal concentrations of H+ and OH− ions, the solution is said to be neutral. If the concentration of H+ is in excess of OH−, the solution is acid, and if less it is alkaline. The ionic product is a known function of the temperature and salt concentrations; hence, if [H+] or [OH−] is known, the other can readily be calculated. For expressing the hydrogen ion concentration, a logarithmic scale is commonly used, where pH is the logarithm of the reciprocal of the hydrogen ion concentration expressed as normality; that is, as equivalents per liter,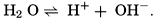
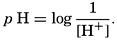 [Equation]. Thus, a neutral solution has a pH of approximately 7, an acid solution a pH less than 7, and an alkaline solution a pH greater than 7. It should be noted that a unit change in pH corresponds to a tenfold change in the hydrogen ion and hydroxyl ion concentrations.
[Equation]. Thus, a neutral solution has a pH of approximately 7, an acid solution a pH less than 7, and an alkaline solution a pH greater than 7. It should be noted that a unit change in pH corresponds to a tenfold change in the hydrogen ion and hydroxyl ion concentrations.The hydrogen ion concentration, or pH, of a solution may be determined in various ways, but all are essentially either electrometric or
Certain organic compounds classed as indicators have the property of changing color over a given range of hydrogen ion concentration. So-called bicolor indicators have one color when in an “acid” solution and another color when in an “alkaline” solution. For any indicator the color change takes place over a definite range in pH, and, at the hydrogen ion concentration that is numerically equal to the dissociation constant of the indicator, equal quantities of both color phases are present in the solution. The range in pH for which various indicators can be used is described by Clark (1928), who also gives in detail the methods for preparing the indicators. For sea water, cresol red and phenol red are generally used, as they cover the pH range normally found in the sea. When working in high pH ranges, bromthymol blue is commonly used.
Three important properties of pH indicators must be known before they can be applied to sea water: the dissociation constant of the indicator, the effect of temperature upon this value, and the salt error. The presence of neutral ions in the solution has a pronounced effect upon the color and, hence, upon the apparent pH as determined by indicators. This is known as the salt error. In general, neutral salts increase the apparent dissociation constant of the indicator, and therefore give low pH readings. In practice a carefully controlled quantity of an indicator solution is added to a sample of sea water, and either the color developed is compared to a set of tubes containing the equivalent quantity of indicator in solutions of known pH or the sample is examined in a bicolorimeter. The accepted colorimetric technique used for determining the pH of sea-water samples and the correction to be applied for salt error and temperature effects are described by Buch (1937) and by Buch and Nynäs (1939). Because of the effects of changes in temperature and pressure upon the dissociation constants of carbonic acid (p. 200) the measured pH of a sea-water sample will differ from the pH in situ. The magnitude of the temperature correction is given by Buch (1937), and the effect of pressure upon the pH has been studied by Buch and Gripenberg (see Buch et al, 1932).
The pH encountered in the sea is between about 7.5 and 8.4. That is, the hydrogen ion concentration ranges from 32 × 10−9 to 4 × 10−9
Alkalinity and Carbon Dioxide Components. The total amount of carbon dioxide in sea water, present either as free gas or bound, may be determined gasometrically after a strong acid has been added to the water to break up the carbonate compounds. Such a method has been described by Greenberg, Moberg, and Allen (1932). In order to determine the carbon dioxide components—namely, the amounts present as carbonic acid (including the free CO2), bicarbonate, and carbonate—titrations must be made. For a given sample of sea water the amount of a strong acid (usually HCl about 0.01 normal) necessary to reduce the pH to about 4.5 is independent of the total CO2. This amount of acid is required to set free the weak acids whose anions have been bound against basic cations. It is therefore not only a measure of the quantity of anions of weak acids in the sample, but also of the cations balanced against them. This quantity, when expressed as the number of milliequivalents of hydrogen ions (mg-atoms of H+) necessary to set free the ions of weak acids in a volume of water which at 20° has a volume of 1 L, is known as the alkalinity. This quantity has also been referred to as the titratable base, excess base, titration alkalinity, and buffer capacity. The term alkalinity has been adopted as the standard designation by the International Association of Physical Oceanography (1939). It should be noted that the term as here defined has no relation to the hydroxyl ion concentration or to the fact that sea water is normally alkaline.
A number of methods have been suggested for determining the alkalinity, and these have been summarised by Thompson and Robinson (1932) and Gripenberg (1937b). In general, they follow one of two techniques. Either the titration is carried out in the presence of the carbon dioxide, in which case the end point is taken at about 4.5, or the carbon dioxide is driven off. In the latter case a higher pH, about
The alkalinity bears a fairly constant relation to the chlorinity. The alkalinity: chlorosity factor for surface water has been determined by a number of workers and found to be close to 0.120 when the alkalinity is expressed in terms of milligram-atoms. The designation specific alkalinity that has been used in some cases is obtained by dividing the alkalinity, as mg-atoms/L, by the chlorinity, in g/kg, but such a mixed ratio should not be used. In water from greater depths the ratio may be somewhat higher than that given above, approaching an upper limit of 0.125 near the sea floor (Wattenberg, 1933). In brackish water the ratio may be increased tremendously if the river water is high in bound carbonate compounds. When the alkalinity: chlorosity factor is to be used as an index in studies of industrial pollution, the “normal” change in the ratio with concentration must first be established in samples of sea water diluted with unpolluted river water. Observations by Moberg and Revelle (1937) and by Wattenberg (1936) have shown that in oceanic water the increase in the Ca:Cl factor with depth is equivalent to the rise in the alkalinity:Cl factor. This indicates that changes in the alkalinity and calcium are of common origin—namely, precipitation or solution of CaCO3. Further material concerning the regional variations in the alkalinity: chlorosity factor is given on p. 208.
Because variations in the alkalinity: chlorosity factor in oceanic water are associated with corresponding changes in the calcium: chlorosity factor, the calcium content of the water may be computed from the alkalinity by the following expression:
This procedure for estimating calcium has been followed by Wattenberg (for example, Wattenberg, 1936).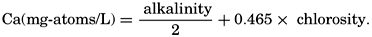
It may be seen that the alkalinity: chlorosity factor of 0.1205 is the same as the HCO3−:Cl factor given in table 35, when the bicarbonate is expressed as milligram-atoms of carbon per liter. The reason for this identity is that, in preparing the table, it was assumed that the pH of the water was such that only bicarbonate ions were present and, hence, would be equivalent to the alkalinity.
In discussions of the carbon dioxide system in sea water, the concentrations of the components have commonly been given in millimoles per liter. These are numerically identical with concentrations given as mg-atoms/L of carbon.
Salts of weak acids containing the following elements are known to occur in sea water: carbon, boron, phosphorus, arsenic, and silicon. Of these, salts of carbonic and boric acid only are present in sufficient concentrations to affect the magnitude of the alkalinity. For the present, we shall neglect the boric acid, which does not affect the alkalinity determination and which has to be considered in the carbonate system only at higher pH's. The alkalinity may then be taken as a measure of the concentration of bicarbonate and carbonate ions, and
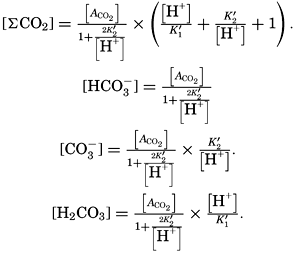
where the brackets indicate molar concentrations—that is, gram-atoms of carbon per liter. The [H2CO3] may be determined by titration with sodium hydroxide and the [CO3−] by titration with acid, using carefully controlled end points. The method is described by Greenberg, Moberg, and Allen (1932). It is shown later that the concentration of either H2CO3 or CO3− will be negligible when the other is present in significant quantities. Therefore we may write: or By substituting in these equations the measured quantities, the other components may be obtained.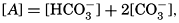
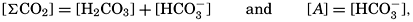
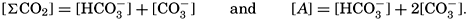
Studies of the carbon dioxide system based on measurements made by these methods have been reported by Greenberg, Moberg, and Allen (1932) and Moberg, Greenberg, Revelle, and Allen (1934). In fig. 38 are shown vertical distribution curves for the carbon dioxide components, total carbon dioxide calculated from the titrations and by direct gasometric measurements, and the alkalinity at a station off the coast of southern California. The total CO2 obtained by the two methods agrees very well and shows a general increase with depth. In the upper layers there is an appreciable amount of CO3−, but this decreases to zero at 200 m, and below this level H2CO3 occurs in quantities which increase with depth. The alkalinity is indicated for the upper 200 m, below which it corresponds, of course, to the curve showing the amount of HCO3−. The increase of alkalinity with depth may be partly due to the biological precipitation of CaCO3 in the upper layers, but in this area it is principally associated with the increasing salinity.
We shall now proceed to a discussion of the laws governing the equilibria between the various carbon dioxide components, the alkalinity,
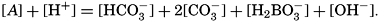
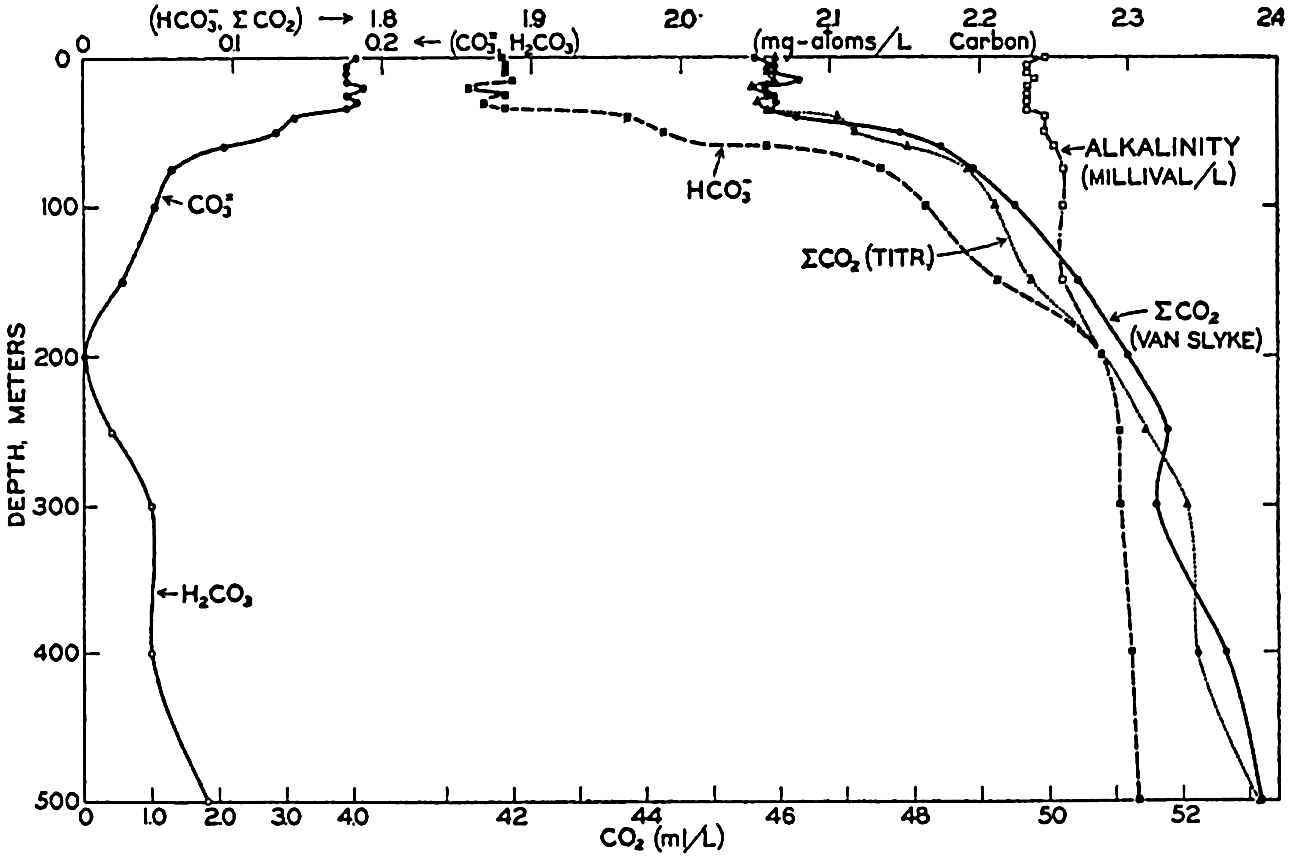
Vertical distribution of the alkalinity and carbon dioxide components off southern California.
[Full Size]
If we indicate the portion of the base (alkalinity) directly bound to the carbon dioxide components as ACO2, the relationship may be written (Buch, 1933a,b)
where all concentrations are in gram-atoms per liter. K′B is the apparent first dissociation constant of boric acid in sea water at the particular temperature and salinity prevailing, and KW is the ionic concentration product of water, [H+] × [OH−], under similar conditions. According to Buch (1938) the ionic product in sea water at 20° can be computed from the following equation: pKW decreases by about 0.035 for each l-degree rise in temperature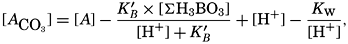
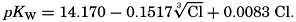
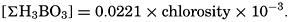
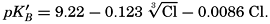
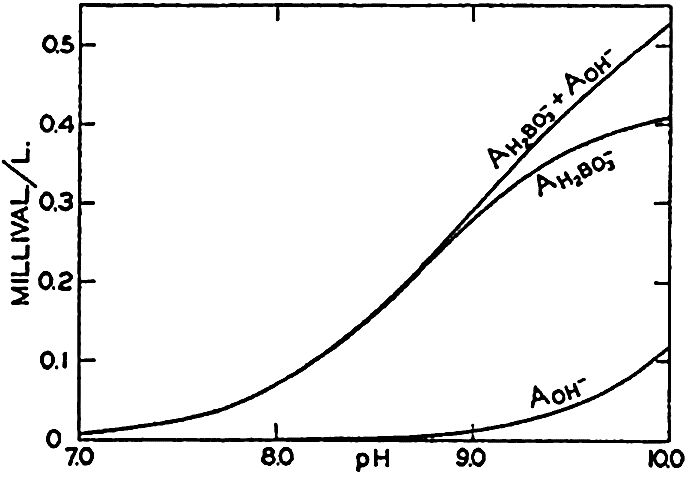
Concentrations of hydroxidebound (AOH−) and borate-bound (AH2BO3−) base as a function of pH in water of Cl = 19.00 ‰ at 20°C.
[Full Size]
The equation for the first dissociation constant of carbonic acid is
and for the second: where the brackets indicate molar concentrations. By introducing the relationships and it is possible to eliminate HCO3− and CO3− from the above equations and obtain them in the following form: Extensive investigations have been carried out to determine the magnitudes of K1′ and K2′. These studies have been reported by Buch, Harvey, Wattenberg, and Gripenberg (1932), and by Moberg, Greenberg, Revelle, and Allen (1934). Buch and others have followed up the work,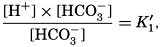
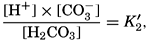
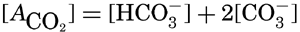
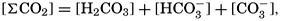
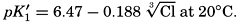
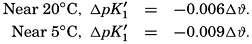
The effect of hydrostatic pressure, as expressed by means of the depth Δz in meters, on the first dissociation constant is
Buch (1938) found that the second dissociation constant of carbonic acid in sea water at 20°C may be computed from the following equation: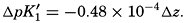
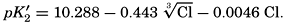
Over the normal range of chlorinity in sea water a simpler expression is adequate—namely,
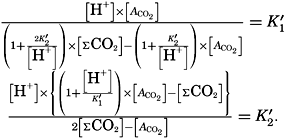
The temperature and pressure corrections are For any given set of conditions, as expressed by means of temperature, salinity, pressure, alkalinity, and pH, the values of ACO2 and K′1 and K′2′ can be calculated. From these the total carbon dioxide and its various components can be computed from the following equations (Revelle, 1934): In fig. 40 are shown the variations in the carbon dioxide components with pH, calculated from the equations given above for sea water of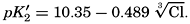
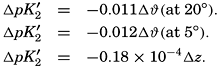
The partial pressure of carbon dioxide is related to the amount of free CO2 + H2CO3 (indicated as H2CO3; see p. 190) in the solution:
The value of cs (p. 191) depends upon the temperature and salinity and the units used for expressing the concentration and partial pressure.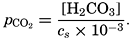
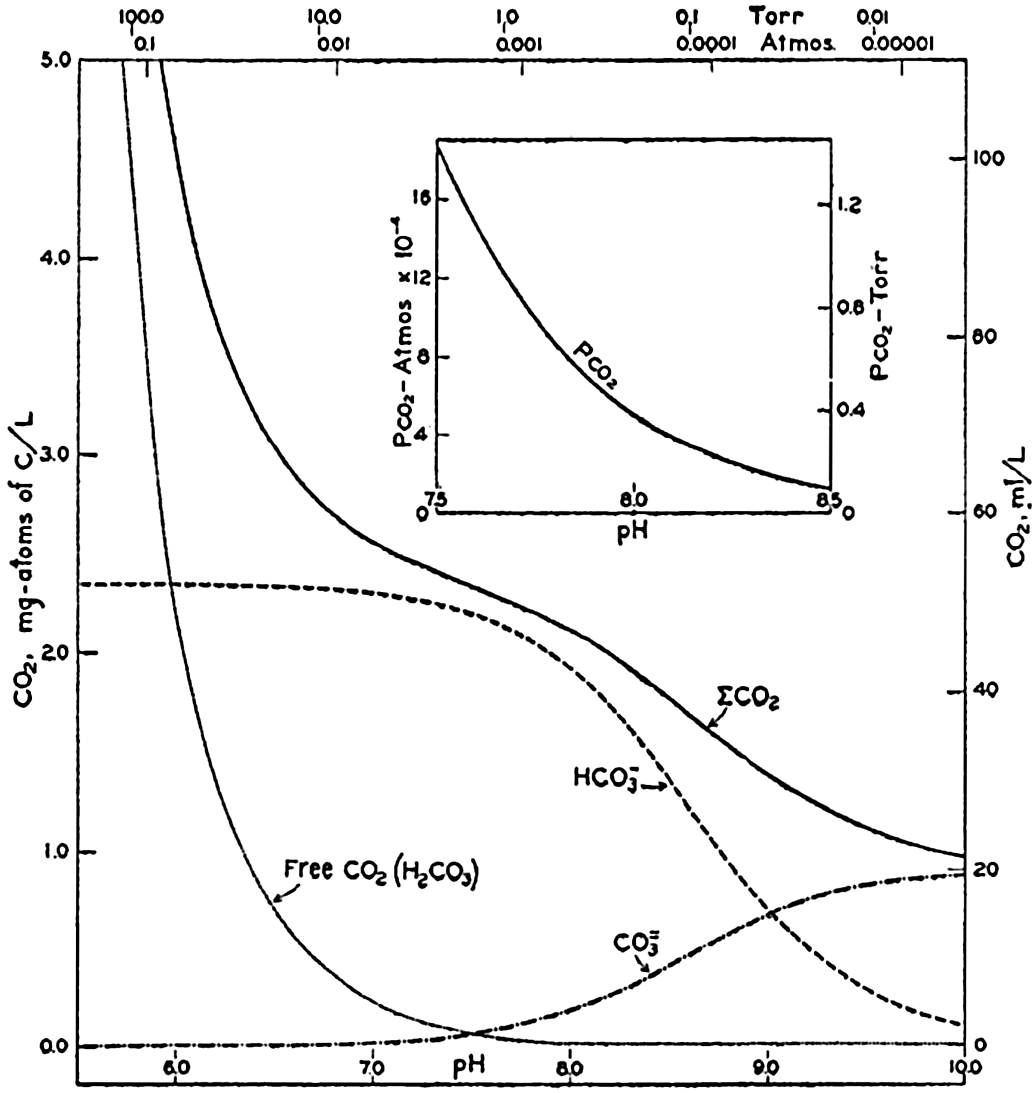
Carbon dioxide components in sea water of Cl = 19.00 ‰ at 20°C as a function of pH and the partial pressure of carbon dioxide.
[Full Size]
In fig. 41 are shown curves for cs at different temperatures and chlorinities, where they represent the amount of H2CO3, in milligram atoms of carbon per liter of sea water, in solution under the designated conditions when the partial pressure of CO2 is 1 physical atmosphere (760 Torr). At 20° and 19 ‰ Cl, cs, is 34.2. That is, a partial pressure of one atmosphere of CO2 would be in equilibrium with a solution containing 34.2 milligram-atoms of carbon as free CO2 + H2CO3. The data are from Buch et al (1932).
The variations in pco2 with the other components is shown in fig. 40. The range is from less than 0.01 to greater than 100 Torr (0.1 × 10−4 to 1000 × 10−4 atm). The relationship over the pH range normally encountered in sea water is shown in the inset diagram in fig. 40. Between pH 7.5 and 8.3, pco2 decreases from 1.4 to 0.15 Torr (18.0 to 2.0 × 10−4 atm). The average partial pressure of CO2 in the air is about 0.23 Torr; hence surface sea water of Cl = 19.0 ‰ at 20° will have a pH of 8.2 if it is in equilibrium with the atmosphere.
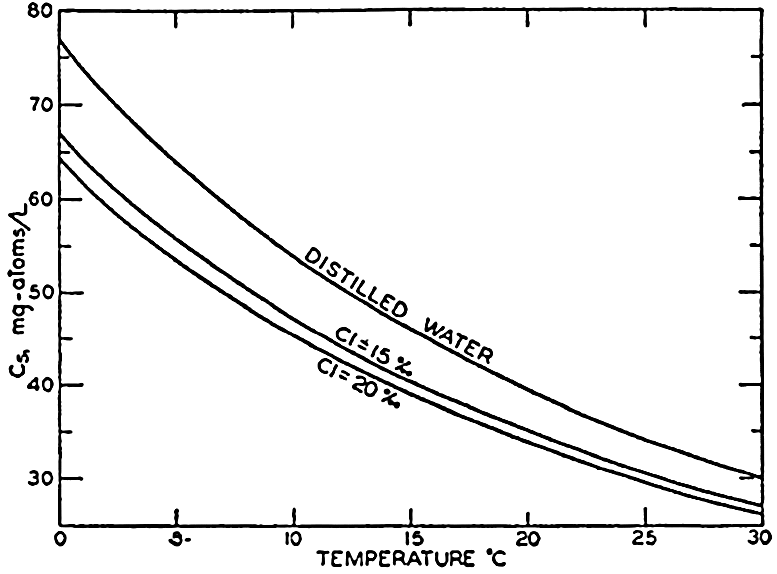
Absorption coefficient (cs) of carbon dioxide in sea water as a function of temperature and chlorinity.
[Full Size]
Sea water is a very favorable medium for the development of photosynthetic organisms. It not only contains an abundant supply of Co2, but removal or addition of considerable amounts results in no marked changes of the partial pressure of CO2 and the pH of the solution, both of which are properties of importance in the biological environment (p. 268). If the CO2 available for photosynthesis is assumed to be H2CO3 + ½HCO3−, 0.48 mg-atoms of carbon per liter may be removed from water of Cl = 19 ‰ with an increase of the pH from 7.5 to only 8.5. In distilled water or in an inert salt solution of zero akalinity initially at pH 7.5, the total CO2 would be about one seventh of that amount.
Buffer Action of Sea Water. If a small quantity of a strong acid or base is added to pure water, there are tremendous changes in the numbers of H+ and OH− ions present, but the changes are small if the acid or base is added to a solution containing a weak acid and its salts or a weak base and its salts. This repression of the change in pH is known as buffer action, and such solutions are called buffer solutions. Sea water contains carbonic and boric acids and their salts and is, therefore, a buffer solution. Let us consider only the carbonate system. Carbonate and bicarbonate salts of strong bases, such as occur in sea water, tend to hydrolyze, and there are always both H+ and OH− ions in the solution. If an acid is added, carbonate is converted to bicarbonate and the bicarbonate to carbonic acid, but, as the latter is a weak acid (only slightly dissociated), relatively few additional hydrogen ions are set free. Similarly, if a strong base is added, the amount of carbonate increases, but the OH− ions formed in the hydrolysis of the carbonate increase only slightly. The buffering effect is greatest when the hydrogen
Cycle of CO2 Between Sea and Atmosphere. Investigations of the partial pressure of CO2 in the ocean and the atmosphere have been made by Krogh (1904) and Buch (1939a,b). The following internal changes will increase or decrease the pCO2 in the surface layer:
| Increase pco2 | Decrease pco2 |
|---|---|
| 1. Rise in temperature | 1. Decrease in temperature |
| 2. Rise in salinity (evaporation) | 2. Decrease in salinity |
| 3. Respiration | 3. Photosynthesis |
| 4. Precipitation of CaCO3 | 4. Solution of CaCO3 |
| 5. Deep water brought to surface |
The partial pressure of CO2 in the surface water can be computed with sufficient accuracy when the temperature, salinity, alkalinity, and pH are known, but, before a better understanding of the CO2 exchange between the sea and the atmosphere can be obtained, a far more comprehensive study of the partial pressure of the atmospheric CO2 must be made. Buch (1939b) has reported a number of direct observations on the CO2 content of the air which indicate that polar air is relatively low in CO2 (pCO2 = 0.23 Torr), compared to continental and tropical air (pCO2 = 0.25 Torr). It has been suggested that in low latitudes the air is enriched with CO2 from the ocean and that the general atmospheric circulation carries the CO2 into high latitudes. There it again dissolves in the sea water, which in time brings it back toward the Equator.
Activity of Ions in Sea Water. The apparent first and second dissociation constants of carbonic acid and the first dissociation constant of boric acid in sea water are larger than in distilled water and increase with increasing salinity. That is, the strength of these acids appears to be greater in solutions containing salts. These phenomena can be accounted for on the theory of activity introduced by Lewis and Randall (1923) and developed mathematically by Debye and Hückel. In a solution containing a mixture of electrolytes, such as sea water, there is a mutual interference of the ions, so that their activity or ability to participate independently in some reaction is much reduced. Most chemical determinations measure the total concentration of some ion and not its activity; however, certain physical measurements show the activity. For example, electromotive force determinations involve the activity of the hydrogen and other ions. Similarly, the measurement of the vapor pressure of a solution of a nonvolatile compound is an indication of the activity of the solvent. The activity will be less than that of the pure solvent under similar conditions. The partial pressure of dissolved gases is a measure of their activity.
The activity coefficient γ is related to the activity α of an ion as in the following example:
Hence, αH+ is the activity of the hydrogen ions expressed as gram-atoms per liter and [H+] is the stoichiometric concentration.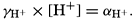
In the studies of the carbon dioxide system in sea water, the total CO2, alkalinity, and carbon dioxide components are measured chemically, and hence the values represent the stoichiometric values and not the activities. On the other hand, the hydrogen ion concentration is determined colorimetrically or electrometrically, and these methods yield the activity of hydrogen ions directly. Therefore, in the equations relating the CO2 components αH+ could have been inserted instead of [H+]. The dissociation constants have been referred to as apparent dissociation constants, in contrast to the thermodynamic constants that would be obtained at infinite dilution, where the activity coefficients (γ) are unity. The apparent dissociation constants are indicated by the symbol prime (′)—for example, K′2. For carbonic acid the thermodynamic second dissociation constant can be written
and this is related to the apparent dissociation constant (Moberg et al, 1934): Similarly, The value of γ H2Co3 depends upon the relative solubility of CO2 in pure water and in sea water at the same temperature and upon the activity of the water in the two cases: The subscript o indicates values at infinite dilution, and the subscript s the values at the concentration under consideration. The values of co and cs can be obtained from fig. 41, and es and eo, (the vapor pressures) can be computed (p. 67).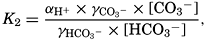
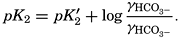
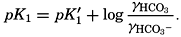
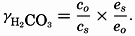
From the empirical equations relating pK′1 and pK′2 to the temperature and chlorinity, we know that at 20° and zero chlorinity the thermodynamic values are pK1 = 6.47 and pK2 = 10.288, and, at 19.00 ‰ Cl, pK'1 = 5.97 and pK'2 = 9.02. By substituting these values and γH2CO3

Empirical equations have been presented which relate pK′1 and pK′2 to the cube root of the chlorinity. It has been shown (Buch et al, 1932, Moberg et al, 1934) that these equations are generally valid for salt solutions other than sea water if the ionic strength is used instead of chlorinity as a measure of concentration. The ionic strength (μ) of a solution is obtained by first multiplying the concentration of each individual type of ion, in moles per kilogram of solvent water, by the square of its valence, and then taking half the sum of these products (Lewis and Randall, 1923, p. 373). Lyman and Fleming (1940) have shown that the ionic strength of sea water in the normal range of concentration may be computed from the expression
Moberg et al (1934) have summarized the pertinent data, but no satisfactory expressions have yet been developed to show the manner in which the activity coefficients of the different ions in sea water change with concentration.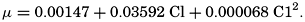
Solubility of CaCO3. The solubility of an electrolyte, such as calcium carbonate, may be expressed by a solubility product. The solubility product is identical with the ionic product (if concentrations are expressed as moles per liter) when the solution is in equilibrium with the solid salt and, therefore, saturated. The value of the solubility product depends upon temperature, the concentration of other ions (salinity), and the hydrostatic pressure. If, under a given set of conditions, the ionic product is less than the solubility product, the solution is undersaturated; if the ionic product is greater the solution is supersaturated, and if suitable nuclei are present, precipitation will proceed until the ionic product equals the solubility product.
The solubility product of CaCO3(KCaCO3) in distilled water at 20° is 5.0 × 10−9. In sea water of chlorinity 19.00 ‰ and at the same temperature, the calcium content is 10.23 mg-atoms/L, and at pH 8.2 the carbonate ion concentration is 0.26 mg-atoms/L of carbon. Therefore the ionic product is
which is 530 times greater than the solubility product in distilled water. If solubility data for distilled water are to be applied to sea water, it is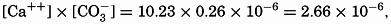
It is therefore necessary to determine empirically the concentrations of Ca++ and CO−3 that can exist in contact with solid CaCO3. Working at 30° and increasing the CO−3 content by lowering the total CO2, Revelle and Fleming (1934) obtained precipitation of CaCO3 as aragonite needles and spherulites. The calcium content of the solution was determined directly, and the CO−3 was calculated from measurements of the pH, alkalinity, and chlorinity. The average of three experiments gave K'CaCO3 = 2.4 × 10−6 at 30°C. Wattenberg has carried out a number of studies on the solubility of calcium carbonate in sea water. His general procedure was to add CaCO3 crystals to sea water, and in some instances to increase the pCO2 The sea-water samples were then placed in sealed flasks and agitated until equilibrium was established. The calcium content of the water was calculated from the alkalinity measurements (p. 196), and the CO3 was obtained with the aid of pH determinations. Wattenberg and Timmerman (1936) give the following values for the apparent solubility product in sea water of Cl = 18.5 to 19.5 ‰:
| Temperature, °C | 0 | 5 | 10 | 15 | 20 | 25 | 30 | 35 |
| K′CaCO3 | 8.1 | 7.9 | 7.4 | 6.8 | 6.2 | 5.5 | 4.7 | 3.8 × 10−7 |
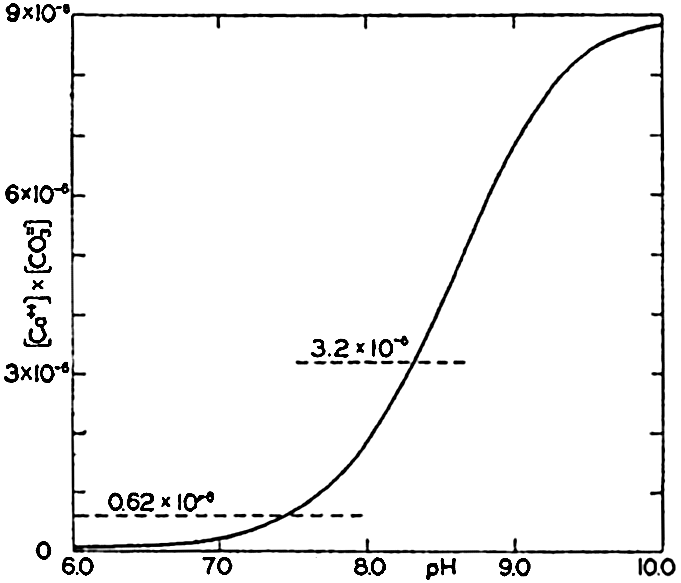
Ionic product [Ca++l × [CO3−] in sea water of Cl = 19.00 ‰ at 20°C as a function of pH. The horizontal lines indicate the solubility product according to Wattenberg (0.62 × 10−6 and according to Revelle and Fleming (3.2 × 10−6).
[Full Size]
At 30° Wattenberg's value is only one fifth of that obtained by Revelle and Fleming. No satisfactory explanation of this difference has yet been offered. In fig. 42 the ionic product in sea water of Cl = 19.00 ‰ at 20° is plotted against pH. The value of K'CaCO3, for this temperature obtained by Wattenberg—namely, 0.62 × l0−6—would indicate that supersaturation exists at all pH's above about 7.5. Surface water in equilibrium with the atmosphere has a pH of about 8.2 and would be
Smith (1940) has investigated the calcium carbonate deposition that takes place in the shallow waters overlying the Bahama Banks. He found the alkalinity to be much reduced, and from measurements he computed the ionic product, [Ca++] × [CO3−]. Minimum values of the product that he considers approach the solubility product were found to fall between the values of Revelle and Fleming and those of Wattenberg.
According to Wattenberg (1936), K'CaCO3 at 20° changes with the chlorinity in the following way:
| Cl, ‰ | 0 | 5 | 10 | 15 | 20 |
| K′CaCO3 | 0.05 | 1.8 | 4.0 | 5.0 | 6.2 × 10−7 |
That is, in sea water the apparent solubility product of CaCO3 increases with chlorinity and decreases with temperature.
Revelle (1934) and Wattenberg (1936) consider that hydrostatic pressure has no significant effect upon the value of K'CaCO3. However, it should be remembered that, because of changes in the dissociation constants of carbonic acid, pressure does modify the relative amounts of HCO3 and CO−3 in the water. Hence, water that is saturated at the surface will be slightly undersaturated if subjected to hydrostatic pressure, even if the total CO2 and calcium contents remain unaltered. Conversely, bottom water saturated with CaCO3 will be supersaturated when brought to the surface.
Although our knowledge of the values of K′CaCO3 under different conditions is incomplete and uncertain, it is possible to show the effect of changes in the conditions upon the ionic product, thus obtaining an understanding of those agencies that will favor precipitation or solution. Revelle (1934) has shown that, in surface water, increase of the salinity and the temperature, and decrease of the pco2—that is, the total CO2 content—all tend to increase the ionic product and therefore favor precipitation. The salinity effect is relatively small, and hence areas of high or rising temperature and of active photosynthesis will be those where precipitation of CaCO3 is most likely to occur. Opposite conditions will favor solution.
In the deep water the range in temperature and salinity is small, and therefore the variations in pCO2 will have the most pronounced effect. According to Wattenberg's studies (1933) the deep waters of the Atlantic Ocean are virtually saturated with calcium carbonate. Areas overlying red clay show slight undersaturation, and those overlying calcareous deposits (globigerina ooze) show either saturation or slight supersaturation.
Certain types of calcareous sedimentary material, both recent and fossil, do not show any evidence of organic origin. These are sometimes considered to be “chemical” deposits. In certain areas, microorganisms undoubtedly play an important part in establishing conditions that result in the incidental precipitation of carbonates. In tropical seas, this process may occur in shoal-water areas, coral reefs, lagoons, and mangrove swamps (Field, 1932). Although microorganisms can produce conditions favoring the precipitation of calcium carbonate, it is considered that they are effective agents in this process only in and on the sediments in the environments listed above. Smith (1940) has found that over the Great Bahama Bank CaCO3 is precipitated, under conditions of heating and excessive evaporation, on nuclei supplied by the sediments. The finely divided CaCO3 in certain deep-sea sediments is thought to arise from the break-down of the shells of foraminifera (p. 982) and not from precipitation in situ.
Distribution of Alkalinity, pH, and Carbon Dioxide Components. In the preceding discussion it has been assumed that the alkalinity: chlorosity factor is constant, and the value 0.1205 (milliequivalents per unit Cl) has been used in computations. This is in agreement with the average of a large number of observations made by Wattenberg (1933) in the Atlantic Ocean and those reported by Revelle (1936) for the upper layers of the Pacific Ocean. Wattenberg's value is commonly given as the specific alkalinity—namely, as milliequivalents per unit chlorinity, in which case it is 0.123. A number of different methods (p. 195) have been employed for measuring alkalinity, but there is no definite proof that they all yield similar values. Hence, it is difficult to compare the results obtained from various parts of the oceans by different workers. However, the findings of Wattenberg (1933), Mitchell and Rakestraw (1933), and Revelle (1936) all show that there is a somewhat higher alka1inity:chlorosity factor in the deeper water than there is in the surface layers. The increase in the factor usually amounts to about 0.005. Wattenberg examined a number of water samples collected immediately over the bottom, and in these he found an even larger factor. The low values in the surface waters are ascribed to removal of calcium carbonate by organisms possessing calcareous skeletons. Smith (1940) has suggested that, at least in certain localities, there may be inorganic precipitation if suitable nuclei are present when
The pH of sea water in contact with the air will vary between about 8.1 and 8.3, depending upon the temperature and salinity of the water and the partial pressure of carbon dioxide in the atmosphere. In areas of great dilution lower values may occur. At subsurface levels, where exchange of carbon dioxide with the atmosphere is impossible, the pH will vary with the extent to which the CO2 content of the water is modified by biological activity. In the euphotic zone, higher pH's are usually found; below this they decrease to a minimum corresponding in general to the layer of minimum oxygen content, and then increase again toward the bottom. Although variations in the salinity affect the pH, the predominant factor is the total carbon dioxide content or its partial pressure.
Unfortunately, differences in technique and in the constants used in arriving at pH's from colorimetric measurements make it difficult to compare the results of different workers. The most extensive studies made on the distribution of pH in the oceans are those of the Meteor (Wattenberg, 1933) in the Atlantic Ocean and those of the Carnegie in the Pacific Ocean. The distributions of pH and dissolved oxygen along a longitudinal profile in the eastern Atlantic Ocean are shown in fig. 43. The rather close similarity of the isolines can easily be seen, and similar patterns would be given by the partial pressure and total CO2 content, although in this case the relations would be inverse ones. The temperature

Distribution of pH and dissolved oxygen in the eastern part of the Atlantic Ocean. The locations of the sections are shown in the map on the left-hand side. The vertical and horizontal scales are different in the two sections. (After Wattenberg.)
[Full Size]
Solubility of Salts in Sea Water
The solubility of calcium carbonate in sea water has been examined in some detail, but relatively little is known about the other constituents. Because of the complex nature of sea water and the effect of other ions upon the activity of any one, the solubility product of a single salt in distilled water cannot be applied to sea water. Cooper (1937b) considers that most of the iron in sea water is not in true solution, but is present in some colloidal form, as the solubility product for the hydroxide is
Thompson and his co-workers (for example, Igelsrud and Thompson, 1936) have carried out extensive phase-rule studies of solutions containing some of the salts in sea water, but so far they have not extended their investigations to natural water.
Some indication of the great solubility of the major constituents is afforded by data on the separation of salts when sea water is frozen (p. 217). Somewhat similar data may be obtained from the evaporation studies by Usiglio (Thompson and Robinson, 1932), which again bring out the fact that sea water is far from saturated with most of the constituents.
| Salt | K Distilled water | K' Sea water S = 35 ‰, ϑ = 20° | Ionic product Cl = 19.0 ‰, ϑ = 20° pH = 8.2 |
|---|---|---|---|
| CaCO3 | 0.5 × 10− 3 | 50 × 10− 3 | 270 × 10− 3 |
| MgCO33H2O | 0.1 × 10− 4 | 3.1 × 10− 4 | 0.14 × 10− 4 |
| SrCO3 | 0.3 × 10− 9 | 500 × 10− 9 | 39 × 10− 9 |
| Mg(OH)2 | 1 × 10− 11 | 5 × 10− 11 | 0.02 × 10− 11 |
The Oxidation-Reduction Potential of Sea Water
The oxidation-reduction potential is a measure of the ability of one chemical system to oxidize a second. It is generally expressed in volts relative to the normal hydrogen electrode. Those substances or solutions having high potentiaIs are able to oxidize those with lower potentials. Although considerable work has been done on the oxidation-reduction potentials in living organisms, little is known about the conditions prevailing in the water. The potential in sea water has been considered
The oxidation-reduction potential of the environment is important to organisms. So-called aerobic bacteria thrive at a higher potential than micro-aerophiles, and anaerobic bacteria can exist only when the potential is low. Hence, in stagnant water and muds where there is no oxygen and the potential is low, only anaerobic forms can exist. The potential is also of geological importance, as the character of certain of the constituents of the sediments will be determined by the prevailing “oxidizing” or “reducing” conditions (p. 996).
Inorganic Agencies Affecting the Composition of Sea Water
The factors that may modify the absolute and the relative concen, trations of the substances in sea water are exchange with the atmosphere-inflow of river water, freezing and melting of sea ice, and biological activity. Biological processes and their effects upon the distribution of various elements are considered in chapter VII.
Exchange with the Atmosphere. The distribution of salinity in the oceans, and hence the concentrations of the major elements, is maintained by agencies that are described elsewhere, but one point must be considered at this time. Over the sea and along its shores, spray is continually being swept up into the air, and as the spray represents actual particles of sea water with its dissolved salts, this process affords a mechanism for the removal of salts from the sea. A large portion of the spray undoubtedly falls back into the water or is carried down by rain (Köhler, 1921). However, winds blowing toward the land will carry with them their content of salt, which may be deposited on the land directly or carried down by the rain. Observations by Jacobs (1937) on the chloride content of the air near the sea showed concentrations ranging between 0.07 and 0.5 mg of chloride per cubic meter of air. The amount increased with the wind velocity and was greatest with onshore winds.
A considerable proportion of the dissolved material carried to the sea by rivers is “cyclic salt”—that is, salt that has been carried inland by the atmosphere and then deposited or carried down by rain and snow (Clarke, 1924, Knopf, 1931).
Besides the exchange of salts that takes place between the atmosphere and the ocean as described above, there is an exchange of dissolved gases and nitrogen compounds which may modify the quantity of these substances present in sea water that is in contact with the atmosphere. The factors that affect the exchange of gases are described elsewhere. The exchange of water between the atmosphere and oceans was taken up in chapter IV.
Rain water contains relatively high concentrations of nitrogen compounds, which are believed to be formed from the constituents of the atmosphere by electrical discharges; hence the atmosphere supplies to the ocean, either directly through rainfall or indirectly through run-off from the land, a certain amount of fixed nitrogen. Whether this increment in the amount of fixed nitrogen is balanced by deposition of organic nitrogen in sediments or by the liberation of gaseous nitrogen through the decomposition of nitrogen compounds in the sea is not yet known.
Effects of Rivers on the Composition of Sea Water. The run-off from land is but a part of the cycle of leaching. The precipitation on the land contains only the cyclic salts, dissolved atmospheric gases, and nitrogen compounds. This water acts upon the rocks, contributing to the mechanical break-down of the solid material and extracting from them their more soluble constituents. The nature and quantity of the various elements dissolved depends upon the character of the rocks or soils with which the water comes in contact on its way to the sea. Because the leaching is carried out by water of low salt concentration yet relatively high in carbon dioxide compounds, it is capable of dissolving materials that would not pass into solution if they were in contact with sea water. In addition to dissolved material, rivers carry to the sea colloidal and particulate material in tremendous quantities. A considerable part of this debris is dropped to the sea bottom near shore, and much of the finer material coagulates and settles when mixed with sea water. Sea water reacts in various ways with the colloidal and finely dispersed material, and some of these reactions may affect the relative composition of the dissolved constituents. Interaction between the dissolved constituents of sea water and the sedimentary debris may be subdivided as follows: (1) solution of the constituents of the sediment, (2) adsorption on the sediment, (3) ionic exchange, and (4) reactions to form new substances. Little is known concerning the importance of these processes.
From the magnitude of the land area drained by rivers emptying into the sea and from the composition of the salts dissolved in river waters,
| Ion | River water (weighted average) | Sea water | River water (less “cyclic” salts) |
|---|---|---|---|
| CO3− | 35.15 | 0.41 (HCO3−) | 35.13 |
| SO4− | 12.14 | 7.68 | 11.35 |
| Cl− | 5.68 | 55.04 | 0.00 |
| NO3− | 0.90 | ............. | 0.90 |
| Ca++ | 20.39 | 1.15 | 20.27 |
| Mg++ | 3.41 | 3.69 | 3.03 |
| Na+ | 5.79 | 30.62 | 2.63 |
| K+ | 2.12 | 1.10 | 2.02 |
| (Fe,Al)2O3 | 2.75 | ............. | 2.75 |
| SiO2 | 11.67 | ............. | 11.67 |
| Sr++, H3BO3, Br− | ...... | 0.31 | ..... |
| 100.00 | 100.00 | 89.75 |
It is not known whether the addition of dissolved solids brings about progressive changes in the relative composition of the sea salts or whether there is any progressive alteration of the total salt content or salinity. In any event, both processes must be exceedingly slow. The total amount of dissolved solids contributed by the rivers each year is only an infinitesimal fraction, 5.4 × 10−8, of the total dissolved solids in the ocean.
| Sea water … … … … … … … … … … … … … … … | 268.45 | 1 |
| Fresh water.… … … … … … … … … … … … … … | 0.1 | “ |
| Continental ice.… … … … … … … … … … … … … | 4.5 | “ |
| Water vapor.… … … … … … … … … … … … … … | 0.003 | “ |
The average composition of the river water is of interest in considering the effect on the oceans as a whole and over long periods, but particular investigations must be concerned with the effects brought about by individual rivers whose dissolved solids may differ markedly in composition and concentration from the average. Data can be obtained from Clarke (1924) or similar sources; as an illustration, values for several of the large American rivers are given in table 44.
From these examples it can be seen that the composition of individual rivers may differ considerably from the average. Thus, the Columbia River is low in chloride and the Colorado River is high, and the latter river is high in sodium and sulphate and below average in calcium and carbonate. The effect that dilution will have upon the chlorosity factors will therefore depend upon the character of the river water.
Thus far we have considered only the more abundant elements in the river water. Undoubtedly, all elements are carried to the sea either in solution or as finely divided particulate material. The high production of plant and animal life which frequently occurs near the mouths of rivers has sometimes been ascribed to the plant nutrients introduced by the rivers. Riley (1937) has found that the Mississippi
Effects of Formation and Melting of Sea Ice on the Composition of Sea Water. A laboratory study of the freezing of sea water was made by Ringer, whose results have been reported by Krümmel (1907) and Johnstone (1928). In these experiments sea water was cooled in the laboratory, and at various temperatures below the initial freezing point the ice and precipitated salts were separated from the mother liquor. Sea water of salinity 35.0‰ begins to freeze at − 1.91°C (p. 66). At first, pure ice crystals separate, and, as the concentration of the brine is increased, the temperature must be further reduced to bring about the formation of additional ice. As the temperature is lowered and the concentration of the brine is increased, the solubility of certain of the dissolved salts is exceeded. At −8.2° the Na2SO4 begins to separate and continues to do so with further cooling. At −23° the NaCl begins to crystallize. In addition, a certain amount of CaCO3 precipitates. Ringer's analyses of the “ice” (including the ice crystals and the precipitated salts) and the brine when the temperature had been reduced to −30° are as follows:
| Ion | Average | Mississippi River | Columbia River | Colorado River |
|---|---|---|---|---|
| CO3− | 35.15 | 34.98 | 36.15 | 13.02 |
| SO4− | 12.14 | 15.37 | 13.52 | 28.61 |
| Cl− | 5.68 | 6.21 | 2.82 | 19.92 |
| NO3− | 0.90 | 1.60 | 0.49 | .......... |
| Ca++ | 20.39 | 20.50 | 17.87 | 10.35 |
| Mg++ | 3.41 | 5.38 | 4.38 | 3.14 |
| Na+ | 5.79 | }8.33* | 8.12 | 19.75 |
| K+ | 2.12 | 1.95 | 2.17 | |
| (Fe,Al)2O3 | 2.75 | 0.58 | 0.08 | .......... |
| SiO2 | 11.67 | 7.05 | 14.62 | 3.04 |
| Annual contribution of dissolved solids (metric tons) | 100,000,000 | 19,000,000 | 13,416,000 | |
| Salt content (g/l) | 0.166 | 0.0924 | 0.702 | |
| * Sum | ||||
One kilogram of sea water, initial salinity 35.05 ‰, yielded:
| Ice crystals ……………………… | 931.9 | g |
| NeCl crystals … … … … … … … | 20.23 | “ |
| Na2SO4 crystals… …………… | 3.95 | “ |
| CaCO3 crystals.… … … … …… | Trace | |
| Brine… … … … ………………… | 43.95 | “ |
The brine contained 23.31 g of H2O and
| Na+……………… | 1.42 g | Cl−……………… | 7.03 g |
| Mg++……………… | 1.31 “ | Br−……………… | 0.08 “ |
| K+……………… | 0.38 “ | SO4−……………… | 0.03 “ |
| Ca++……………… | 0.39 “ |
From these data it is readily seen that, when the temperature of the ice and brine is lowered to −30°, there are marked differences in the relative composition of the salts in the “ice” and in the brine. If the cooling is continued to −50°, there is further separation of ice and salt crystals, but some very concentrated brine is still present.
From these experiments it would appear that the formation of sea ice might have a pronounced effect upon the relative composition of the salts in the water. The brine from which it formed would be modified in one direction, and, if melting took place in water other than that from which the ice formed, the effect would be in the opposite direction. However, the formation of sea ice in nature is not reproduced by these laboratory experiments. Let us suppose that, in a region where the depth to the bottom is moderate or great, sea water of normal composition is subjected to cooling at the surface. The resulting increase in density gives rise to convection movements that continue until the water at the surface reaches the freezing point, and then ice will begin to form. The brine, being of greater concentration but having virtually the same temperature, will sink and new water will be brought toward the surface and into contact with the ice. At first, isolated, elongated ice crystals are produced, but as the freezing continues these form a matrix in which a certain amount of the brine is mechanically included. The ice crystals themselves are at this stage probably “pure ice.” If the freezing proceeds rapidly, the brine will accumulate in separate cells within the body of the ice and, as the temperature of the ice near the surface is reduced, more ice crystals are formed, the cells decrease in size, and the concentration of the brine in the cells increases (fig. 16, p. 72). This may continue so far that solid salts crystallize in the cells. From this it can be seen that there is not necessarily any relative change in the composition of the dissolved salts in the sea water and in the sea ice (ice crystals plus the enclosed brine).
The salinity of the ice, using the same definition as applied to sea water, has been shown to depend upon the rate of freezing. Malmgren (1927), from the observations made by the Maud Expedition, gives the
| Air temperature (°C) | Salinity of ice (‰) |
| − 16 | 5.64 |
| − 28 | 8.01 |
| − 30 | 8.77 |
| −40 | 10.16 |
The salinities are based upon chlorinity determinations made on samples of the melted ice. The salinity of the surface water was about 30 ‰. The effect of the rate of freezing is also shown by the analyses of samples obtained in April from an ice floe that had started to form the preceding November:
| Distance from surface of ice (cm) | 0 | 6 | 13 | 26 | 45 | 82 | 95 |
| Salinity of ice (‰) | 6.74 | 5.28 | 5.31 | 3.84 | 4.37 | 3.48 | 3.17 |
The lower salt content of the deeper ice is related to the slower rate of formation. When ice is formed with extreme rapidity, its salinity will approach that of the water from which it is produced.
According to Ringer's experiments the cooling of ice containing cells of brine leads to formation of additional ice crystals and, if the temperature is reduced sufficiently, to separation of salt crystals within the ice. With very rapid freezing, brine and salt crystals may accumulate on the surface of the ice, making the surface “wet” at temperatures of −30° to −40°C and greatly increasing the friction against sled runners or skis.
In such rapidly frozen ice the cells containing the brine are large or numerous. If the temperature rises, the ice surrounding thg cells melts and the separated salt crystals are dissolved, but before complete solution has taken place the brine cells may join, permitting the brine to trickle through the ice. Under these conditions some of the solid salts may be left in the ice, and the composition of the water obtained by melting will differ from that of normal sea water. If, on the other hand, the temperature of the ice is raised to 0°C, all salts dissolve, the cells grow so large that the ice becomes porous, all brine trickles down from the portions of the ice above the sea surface, and the exposed old ice becomes fresh and can be used as a source of potable water.
Analyses by Wiese (1930) indicate that the processes that have been described may be effective in changing the relative composition of the salts, He found that the sulphate and alkalinity factors were greater in the ice than in the water and were greater in old ice than in newly frozen ice. This indicates that small amounts of sulphate, probably present as Na22SO4, have remained in the ice during the process of ageing and, probably, that the relative amounts of CaCO3 have changed.
Results of the Maud Expedition, reported by Malmgren (1927) and Sverdrup (1929), are not in agreement with the findings of Wiese. Chlorinities of water obtained by melting ice were systematically higher when determined by titration than when computed by means of Knudsen's Hydrographical Tables from observations of density. This discrepancy was interpreted to mean that the sea ice contains an excess of chlorides, but it may arise from the application of Knudsen's Tables to water that has been diluted by essentially distilled water, as explained on p. 59. The fact that the SO4/Cl ratio was nearly the same in the ice and the sea water (Malmgren, 1927, p. 9) supports the latter explanation and indicates that no changes in relative concentration had resulted from processes of freezing and melting. The problem, however, cannot be considered solved, and it offers opportunities for further laboratory investigations and observations in the field.
Geochemistry of the Ocean Waters
The total quantity of dissolved solids present in the waters of the oceans can be estimated by assuming an average salinity of 35 ‰ and assuming that the volume of the ocean is 1.37 × 109 km3 (p. 15). With a density in situ of 1.04 for the ocean waters, the dissolved solids amount to 5 × 1016 metric tons. This immense quantity of material would form a layer of dried salts 45 m thick over the entire earth, or 153 m thick over the present land area. The amount in tons of any element may be estimated by multiplying the value given in the first column in table 45 by 1.42 × 1012. The figures for the variable elements correspond to the higher values listed in table 36. Obviously the total amounts of even the trace elements are tremendous, and, if methods of extraction were economically feasible, the oceans would serve as an “inexhaustible” source of these substances.
| Element | Sea water S = 35‰ (mg/kg) | Potential “supply” in 600 g of rock (mg/kg of sea water) | Percentage in solution |
|---|---|---|---|
| Silicon | 4 | 165,000 | 0.002 |
| Aluminum | 0.5 | 53,000 | 0.001 |
| Iron | 0.02 | 31,000 | 0.0001 |
| Calcium | 408 | 22,000 | 1.9 |
| Sodium | 10,769 | 17,000 | 65 |
| Potassium | 387 | 15,000 | 2.6 |
| Magnesium | 1,297 | 13,000 | 10 |
| Titanium | ............ | 3,800 | ? |
| Manganese | 0.01 | 560 | 0.002 |
| Phosphorus | 0.1 | 470 | 0.02 |
| Carbon | 28 | 300 | 9 |
| Sulphur | 901 | 300 | 300 |
| Chlorine | 19,353 | 290 | 6700 |
| Strontium | 13 | 250 | 5 |
| Barium | 0.05 | 230 | 0.02 |
| Rubidium | 0.2 | 190 | 0.1 |
| Fluorine | 1.4 | 160 | 0.9 |
| Chromium | p | 120 | ? |
| Zirconium | ............ | 120 | ? |
| Copper | 0.01 | 60 | 0.02 |
| Nickel | 0.0001 | 60 | 0.0002 |
| Vanadium | 0.0003 | 60 | 0.0005 |
| Tungsten | ............ | 41 | ? |
| Lithium | 0.1 | 39 | 0.2 |
| Cerium | 0.0004 | 26 | 0.002 |
| Cobalt | p | 24 | ? |
| Tin | p | 24 | ? |
| Zinc | 0.005 | 24 | 0.02 |
| Yttrium | 0.0003 | 19 | 0.002 |
| Lanthanum | 0.0003 | 11 | 0.003 |
| Lead | 0.004 | 10 | 0.04 |
| Molybdenum | 0.0005 | 9 | 0.005 |
| Thorium | <0.0005 | 6 | 0.01 |
| Cesium | 0.002 | 4 | 0.05 |
| Arsenic | 0.02 | 3 | 0.7 |
| Scandium | 0.00004 | 3 | 0.001 |
| Bromine | 66 | 3 | 2000 |
| Boron | 4.7 | 2 | 240 |
| Uranium | 0.015 | 2 | 0.8 |
| Selenium | 0.004 | 0.4 | 1 |
| Cadmium | p | 0.3 | ? |
| Mercury | 0.00003 | 0.3 | 0.001 |
| Iodine | 0.05 | 0.2 | 25 |
| Silver | 0.0003 | 0.06 | 0.5 |
| Gold | 0.056 | 0.003 | 0.3 |
| Radium | 0.093 | 0.066 | 0.05 |
| p = present | |||
According to present theories, most of the solid material dissolved in the sea originated from the weathering of the crust of the earth. The problem as to the amount of rock weathered has been treated by Goldschmidt (1933) in the following way. For each square centimeter of the surface of the earth there are 278 kg of sea water; therefore, for each square centimeter the ocean water contains very nearly 3 kg of sodium. The average sodium content of igneous rocks is 2.83 per cent, and in sedimentary deposits it is 1.00 per cent. In the process of weathering, a certain amount of the material is leached away, and Goldschmidt estimates that the mass of the sedimentary deposits (Y) is 0.97 of the original igneous rocks (X) that gave rise to them. Therefore,
From this we find that for each square centimeter of the earth's surface about 160 kg of igneous rocks have been weathered. Therefore, approximately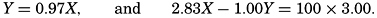
Examination of table 45 shows that the elements may be grouped in three classes, depending upon the percentage in solution: (1) Sulphur, chlorine, bromine, and boron occur in amounts greater than those which could have been supplied by the weathering of the 600 g of rock. Goldschmidt considers that these elements were present in the primeval atmosphere as volatile compounds and that they accumulated in the ocean waters in the earliest times. (2) Calcium, sodium, potassium, magnesium, carbon, strontium, selenium, and iodine, which form relatively soluble compounds, are present in sea water in amounts greater than 1 per cent of the potential supply. (3) The remaining elements, which are present in small amounts.
It is striking that silicon, aluminum, and iron, the most abundant elements in igneous rocks (oxygen is actually the most abundant, but does not have to be considered here), are present in sea water in extremely small amounts. Thus, the relative abundance of the elements in sea water differs markedly from that in the earth's crust. With a few exceptions, all of the elements have been potentially available in much larger amounts than are actually present in solution. The relative composition of river water differs from that of sea water, and, in addition to the dissolved constituents, rivers introduce large quantities of particulate material that would pass into solution if the sea water were unsaturated with respect to these substances. Therefore, it appears that factors operating in the sea itself must control the concentrations of many of the elements that are potentially available in Iarge amounts. These factors are solubility, physical-chemical reactions, and biological activity. Our present knowledge is inadequate to designate which process or processes may control the concentration of a given element. Therefore, the following remarks will merely indicate the character of the factors that may be involved.
Certain elements may be present in such amounts that the solubility of their compounds may limit their concentration. In these eases,
Bibliography
Atkins, W. R. G.1923. The phosphate content of fresh and salt waters in its relationship to the growth of algal plankton. Marine Biol. Assn. U. K., Jour., v. 13, p. 119–50, 1923. Plymouth. 
Atkins, W. R. G.1936.
“Estimation of zinc in sea water using sodium diethyldithiocarbamate”
. Marine Biol. Assn. U. K., Jour., v. 20, p. 625–26, 1936. Plymouth.
Ball, Eric G., and C. C. Stock. 1937.
“The pH of sea water as measured with the glass electrode”
. Biol. Bull., v. 73, p. 221–26, 1937.
Bein, Willy, H. Hirsekorn, L. Möller. 1935. Konstantenbestimmungen des Meerwassers und Ergebnisse über Wasserkörper. Berlin, Universität, Institut f. Meereskunde, Veröff., N.F., A. Geogr.-naturwiss. Reihe, Heft 28, 240 pp., 1935.
Boury, M.1938. Le plomb dans le milieu marin. L'Office des Pêches maritimes. Revue des Travaux scientifiques, v. 11, p. 157–66, 1938. Paris.
Buch, Kurt. 1933a. Der Borsäuregehalt des Meerwassers und seine Bedeutung bei der Berechnung des Kohlensäuresystems im Meerwasser. Conseil Perm. Internat. p. l'Explor. de la Mer, Rapp. et Proc.-Verb., v. 85, p. 71–75, 1933.
Buch, Kurt. 1933b.
“On boric acid in the sea and its influence on the carbonic acid equilibrium”
. Conseil Perm. Internat. p. l'Explor. de la Mer, Jour. du Conseil, v. 8, p. 309–25, 1933.
Buch, Kurt. 1937. Die kolorimetrische Bestimmung der Wasserstoffionenkoncentration im Seewasser. pt. 2, p. 27–33 in: Wattenberg, H., Critical review of the methods used for determining nutrient salts and related constituents in salt water. Conseil Perm. Internat. p. l'Explor. de la Mer, Rapp. et Proc.-Verb., v. 103, 1937.
Buch, Kurt. 1938.
“New determination of the second dissociation constant of carbonic acid in sea water”
, Acta Acad. Aboensis, Math. et Physica, v. 11, no. 5, 18 pp. 1938. Abo, Finland.
Buch, Kurt. 1939a.
“Beobachtungen über das Kohlensäuregleichgewicht und über den Kohlensäureaustausch zwischen Atmosphäre und Meer im Nord-Atlantischen Ozean”
. Acta Acad. Aboensis, Math. et Physica, v. 11, no. 9, 32 pp., 1939. Abo, Finland.
Buch, Kurt. 1939b.
“Kohlensäure in Atmosphäre und Meer an der Grenze zum Arktikum”
. Acta Acad. Aboensis, Math. et Physica, v. 11, no. 12, 41 pp., 1939. Abo, Finland.
Buch, Kurt, H. W. Harvey, H. Wattenberg, and S. Gripenberg. 1932. Über das Kohlensäuresystem im Meerwasser. Conseil Perm. Internat. p. l'Explor. de la Mer, Rapp. et Proc.-Verb., v. 79, 70 pp., 1932.
Buch, Kurt, and Ole Nynäs.1939.
“Studien Über neuere pH-Methodik mit besonderer Berücksichtigung des Meerwassers”
. Acta Acad. Aboensis, Math. et Physica, v. 12, no. 3, 41 pp., 1939. Abo, Finland.
Clark, W. M.1928. The determination of hydrogen ions. 3rd ed., Baltimore, Williams and Wilkins, 717 pp., 1928.
Clarke, F. W.1924.
“The data of geochemistry”
. 5th ed.U. S. Geol. Survey, Bull.no. 770, 841 pp., 1924. Washington, D. C.
Closs, Karl. 1931. Über das Vorkommen des Jods im Meer und in Meeresorganismen. Morten Johansen, Oslo. 150 pp., 1931.
Clowes, A. J.1938. Phosphate and silicate in the southern oceans. Discovery Repts., v. 19, p. 1–120, 1938.
Cooper, L. H. N.1935.
“Iron in the sea and in marine plankton”
. Roy. Soc., Proc., Ser. B, v. 118, p. 419–38, 1935. London.
Cooper, L. H. N.1937a.
“Oxidation-reduction potential in sea water”
. Marine Biol. Assn. U. K., Jour., v. 22, p. 167–76, 1937. Plymouth.
Cooper, L. H. N.1937b.
“Some conditions governing the solubility of iron”
. Roy. Soc., Proc., Ser. B, v. 124, p. 299–307, 1937. London.
Cooper, L. H. N.1938a.
“Salt error in determinations of phosphate in sea water”
. Marine Biol. Assn. U. K., Jour., v. 23, p. 171–78, 1938. Plymouth.
Cooper, L. H. N.1938b.
“Redefinition of the anomaly of the nitrate-phosphate ratio”
. Marine Biol. Assn. U. K., Jour., v. 23, p. 179, 1938. Plymouth.
Dietz, R. S., K. O. Emery, and F. P. Shepard. 1942.
“Phosphorite deposits on the sea floor off southern California”
. Geol. Soc. Amer., Bull, v. 53, p. 815—48, 1942.
Dittmar, W.1884. Report on researches into the composition of ocean water, collected by H.M.S. Challenger. Challenger Repts., Physics and Chem., v. 1, p. 1–251, 1884.
Dorsey, N. Ernest. 1940.
“Properties of ordinary water-substance”
. Amer. Chem. Soc., Monograph Ser.No. 81, New York, Reinhold Pub. Corp., 673 pp., 1940.
Ernst, Theodor, and Hans Hoermann. 1936. Bestimmung von Vanadium, Nickel und Molybdän im Meerwasser. Gesellsch. d. Wiss. zu Göttingen.
Evans, R. D., A. F. Kip, and E. G. Moberg. 1938.
“The radium and radon content of Pacific Ocean water, life, and sediments”
. Amer. Jour. Sci., v. 36, p. 241–59, 1938.
Field, R. M.1932.
“Microbiology and the marine limestones”
. Geol. Soc. Amer., Bull., v. 43, p. 487–93, 1932.
Fox, C. J. J.1907. On the coefficients of absorption of the atmospheric gases in distilled water and sea water. Conseil Perm. Internat. p. l'Explor. de la Mer, Pub. de Circonstance, no. 41, 27 pp., 1907.
Fox, C. J. J.1909.
“On the coefficients of absorption of nitrogen and oxygen in distilled water and sea water and of atmospheric carbonic acid in sea water”
. Faraday Soc., Trans., v. 5, p. 68–87, 1909.
Fox, H. Munro, and Hugh Ramage. 1931.
“A spectrographic analysis of animal tissues”
. Roy, Soc., Proc., Ser. B, v. 108, p. 157–73, 1931. London.
Föyn, Ernst, B. Karlik, H. Pettersson, and E. Rona. 1939. The radioactivity of seawater. Oceanografiska Inst. Göteborg (Göteborgs K. Vetensk… … Handlingar, 5, Ser. B), Meddelanden, N. S., 44 pp., 1939.
Gaarder, T.1916. De vestlandske fjordes hydrografi. I. Surstoffet i fjordene. Bergens Mus. Aarbok, 1915–16.
Goldschmidt, V. M.1933. Grundlagen der quantitativen Geochemie. Fortschritte der Mineral., Kristal. und Petrographie, v. 17, p. 112–56, 1933.
Goldschmidt, V. M.1937.
“The principles of distribution of chemical elements in minerals and rocks”
. Chem. Soc., Jour., p. 655–73, 1937. London.
Goldschmidt, V. M., and L. W. Strock. 1935. Zur Geochemie des Selens, II. Gesellsch. d. Wiss. zu Göttingen, Math.-Phys. Klasse. Fachgruppe IV, Geol. u. Mineral., N.F., v. 1, p. 123–42, 1935.
Greenberg, D. M., E. G. Moberg, and E. C. Allen. 1932.
“Determination of carbon dioxide and titratable base in sea water”
. Ind. Eng. Chem., Anal. ed., v. 4, p 309–13, 1932.
Gripenberg, Stina. 1937a.
“The calcium content of Baltic water”
. Conseil Perm. Internat. p. l'Explor. de la Mer, Jour. du Conseil, v. 12, p. 293–304, 1937.
Gripenberg, Stina. 1937b.
“The determination of excess base in seawater”
. Internat. Assn. Phys. Oceanogr. (Assn. d'Océanogr. Phys.), Union Géod. et Géophys. Internat., Proc.-Verb., no. 2, p. 150–52, 1937. Liverpool.
Haber, F.1928. Das Goldim Meere. Zeitschr. d. Gesellsch. f. Erdkunde, Suppl. 3, p. 3–12, 1928.
Haendler, H. M., and T. G. Thompson. 1939.
“The determination and occurrence of aluminum in sea water”
. Jour. Marine Research, v. 2, p. 12–16, 1939.
Harding, M. W., and E. G. Moberg. 1934. Determination and quantity of boron in sea water. Fifth Pacific Sci. Cong., Canada, 1933, Proc., v. 3, p. 2093–95, 1934.
Harvey, H. W.1926.
“Nitrates in the sea”
. Marine Biol. Assn. U. K., Jour., v. 14, p. 71–88, 1926. Plymouth.
Harvey, H. W.1937.
“The supply of iron to diatoms”
. Marine Biol. Assn. U. K., Jour., v. 22, p. 205–19, 1937. Plymouth.
Hewitt, L. F.1937. Oxidation-reduction potentials in bacteriology and biochemistry. 4th ed. London County Council, no. 3200, 101 pp., 1937.
Igelsrud, Iver, and T. G. Thompson. 1936.
“Equilibria in the saturated solutions of salts occurring in sea water. II”
. The quaternary system MgCl2− CaCI2−KC1-H2O at 0° Amer. Chem. Soc., Jour., v. 58, p. 1–13, 1936.
Igelsrud, Iver, T. G. Thompson, and B. M. G. Zwicker. 1938.
“The boron content of sea water and of marine organisms”
. Amer. Jour. Sci., v. 35, p. 47–63, 1938.
International Assn. Phys. Oceanography. (Assn. d'Océanographie Physique, Union Géodesique et Géophysique Internationale.) Report of the Committee on Chemical Methods and Units. Presented at 7th General Assembly, Washington, D. C., 1939. Publication scientifique. (In press.)
Jacobs, Woodrow C.1937.
“Preliminary report on a study of atmospheric chlorides”
. Monthly Wea. Review, v. 65, p. 147–51, 1937. Washington, D, C.
Jacobsen, J. P., and Martin Knudsen. 1940.
“Urnormal 1937 or primary standard sea-water 1937”
. Internat. Assn. Phys. Oceanogr. (Assn. d'Océanogr. Phys., Union Géod. et Géophys. Internat.) Pub. sci. 7, 38 pp., 1940. Liverpool.
Johnstone, James. 1928. An introduction to oceanography. Liverpool, University Press, 368 pp., 1928.
Kirk, P. L., and E. G. Moberg. 1933.
“Microdetermination of calcium in sea water”
. Ind. Eng. Chem., Anal. ed., v. 5, p. 95–97, 1933.
Knopf, A.1931.
“Age of the ocean. Physics of the earth”
, v. 4, Age of the earth, pt. 2, p. 65–72. Nat. Res. Council, Bull., no. 80, 1931. Washington, D. C.
Köhler, Hilding. 1921. Zur Kondensation des Wasserdampfes in der Atmosphäre. Geofysiske Publikasjoner, v. 2, no. 1, 15 pp., 1921. Oslo.
Krogh, August. 1904.
“On the tension of carbonic acid in natural waters and especially in the sea”
. Medd. om Grönland, v. 26, p. 342, 1904.
Krogh, August. 1934.
“A method for the determination of ammonia in water and air”
. Biol. Bull., v. 67, p. 126–131, 1934.
Krümmel, Otto. 1907. Handbuch der Ozeanographie. Bd. 1. Die räumlichen, chemischen und physikalischen Verhältnisse des Meeres. Stuttgart, J. Engelhorn, 526 pp., 1907.
Lewis, G. N., and Merle Randall. 1923. Thermodynamics and the free energy of chemical substances. N. Y., McGraw-Hill, 653 pp., 1923.
Lyman, John, and R. H. Fleming. 1940.
“Composition of sea water”
. Jour. Marine Research, v. 3, p. 134–46, 1940.
McClendon, J. F., C. C. Gault, and S. Mulholland. 1917. The hydrogen-ion concentration, CO2− tension, and CO2−content of sea water. Carnegie Inst.Washington, Pub. no. 251, Papers from Dept. Marine Biol., p. 21–69, 1917.
Malmgren, Finn. 1927.
“On the properties of sea-ice”
. Norwegian North Polar Exped. with the Maud 1918–1925, Sci. Results, v. 1, no. 5, 67 pp., 1927.
Marks, Graham. 1938.
“The copper content and copper tolerance of some species of mollusks of the southern California coast”
. Biol. Bull., v. 75, p. 224–37, 1938.
Michaelis, L.1930.
“Oxidation-reduction potentials”
. Phila., Lippincott, 199 pp., 1930.
Mitchell, P. H., and N. W. Rakestraw. 1933.
“The buffer capacity of sea water”
. Biol. Bull., v. 65, p, 437–451, 1933.
Moberg, E. G., D. M. Greenberg, R. Revelle, and E. C. Allen. 1934.
“The buffer mechanism of sea water”
. Scripps Inst. Oceanogr., Calif. Univ., tech. ser., v. 3, p. 231–78, 1934.
Moberg, E. G., and R. R. D. Revelle. 1937.
“The distribution of dissolved calcium in the North Pacific”
. Internat. Assn. Phys. Oceanogr. (Union Géod. et Géophys. Internat., Assn. d'Océanogr. Phys.), Procès-verb., no. 2, p. 153, 1937.
Rakestraw, Norris W.1936.
“The occurrence and significance of nitrite in the sea”
. Biol. Bull., v. 71, p. 131–67, 1936.
Rakestraw, N. W., and V. M. Emmel. 1937.
“The determination of dissolved nitrogen in water”
. Ind. Eng. Chem., Anal. ed., v. 9, p. 344–46, 1937.
Rakestraw, N. W., and V. M. Emmel. 1938a.
“The relation of dissolved oxygen to nitrogen in some Atlantic waters”
. Jour. Marine Research, v. 1, p. 207–16, 1938.
Rakestraw, N. W., and V. M. Emmel. 1938b.
“The solubility of nitrogen and argon in sea water”
. Jour. Phys. Chem., v. 42, p. 1211–15, 1938.
Rakestraw, N. W., C. E. Herrick, Jr., and W. D, Urry. 1939.
“The helium-neon content of sea water and its relation to the oxygen content”
. Amer. Chem. Soc., Jour., v. 61, p. 2806–07, 1939.
Rakestraw, N. W., and F. B. Lutz. 1933.
“Arsenic in sea water”
. Biol. Bull., v. 65, p. 397–401, 1933.
Rakestraw, N. W., H. E. Mahncke, and E. F. Beach. 1936.
“Determination of iron in sea water”
. Ind. Eng. Chem., Anal. ed., v. 8, p. 136–38, 1936.
Reith, J. F.1930.
“Der Jodgehalt von Meerwasser”
. Recueil Trav. chim. Pays-Bas, v. 49, p. 142–50, 1930.
Revelle, Roger. 1934.
“Physico-chemical factors affecting the solubility of calcium carbonate in sea water”
. Jour. Sedim. Petrol., v. 4, p. 103–10, 1934.
Revelle, Roger. 1936. Marine bottom samples collected in the Pacific Ocean by the Carnegie on its seventh cruise. California University, Dissertation, 1936. (Carnegie Inst.Washington, Carnegie Repts. In press.)
Revelle, Roger, and R. H. Fleming. 1934.
“The solubility product constant of calcium carbonate in sea water”
. Fifth Pacific Sci. Cong., Canada, 1933, Proc., v. 3, p. 2089–92, 1934.
Riley, G. A.1937.
“The significance of the Mississippi River drainage for biological conditions in the northern Gulf of Mexico”
. Jour. Marine Research, v. 1, p. 60–74, 1937.
Robinson, Rex J., and F. W. Knapman. 1941.
“The sodium-chlorinity ratio of ocean waters from the northeast Pacific”
. Jour. Marine Research, v. 4, p. 142–152, 1941.
Robinson, Rex J., and H. E. Wirth. 1934. Report on the free ammonia, albuminoid nitrogen and organic nitrogen in the waters of the Puget Sound area, during the summers of 1931 and 1932. Conseil Perm. Internat. p. l'Explor. de la Mer, Journal du Conseil, v. 9, p. 15–27, 1934.
Robinson, Rex J., and H. E. Wirth. 1935.
“Photometric investigation of the ceruleomolybdate determination of phosphate in waters”
. Ind. Eng. Chem., Anal. ed., v. 7, p. 147–50, 1935.
Rogers, C. G.1938. Textbook of comparative physiology. 2d ed.New Pork, McGraw-Hill, 715 pp., 1938.
Smith, C. L.1940.
“The Great Bahama Bank. II. Calcium carbonate precipitation”
. Jour. Marine Research, v. 3, p 171–189, 1940.
Ström, K. M.1936. Land-locked waters. Hydrography and bottom deposits in badly ventilated Norwegian fjords with remarks upon sedimentation under anaerobic conditions. Norske Vidensk. Ak. i Oslo, 1. Math.-Naturv. Klasse, no. 7, 85 pp., 1936.
Subow, N. N.1931.
“Oceanographical tables. U.S.S.R.”
, Oceanogr. Institute, Hydro-meteorol. Com., 208 pp., 1931. Moscow.
Sverdrup, H. U.1929.
“The waters on the North-Siberian Shelf. Norwegian North Polar Exped. with the Maud 1918–1925”
, Sci. Results, v. 4, no. 2, 206 pp., 1929.
Thomas, Bertram D., and T. G. Thompson. 1933.
“Lithium in sea water”
. Science, v. 77, p. 547–48, 1933.
Thompson, T. G., and R. W. Bremner. 1935a.
“The determination of iron in sea water”
. Conseil Perm. Internat. p. l'Explor. de la Mer, Jour. du Conseil, v. 10, p. 33–38, 1935.
Thompson, T. G., and R. W. Bremner. 1935b. The occurrence of iron in the water of the northeast Pacific Ocean. Conseil Perm. Internat. p. l'Explor. de la Mer, Jour. du Conseil, v. 10, p. 39–47, 1935.
Thompson, T. G., and H. G. Houlton. 1933.
“Determination of silicon in sea water”
. Ind. Eng. Chem., Anal, ed., v. 5, p. 417–21, 1933.
Thompson, T. G., W. R. Johnston, and H, E. Wirth. 1931. The sulfate-chlorinity ratio in ocean water. Conseil Perm. Internat. p. l'Explor. de la Mer, Jour. du Conseil, v. 6, p. 246–51, 1931.
Thompson, T. G., and R. J. Robinson. 1932. Chemistry of the sea. Physics of the earth, v. 5, Oceanography, p. 95–203. Nat. Research Council, Bull., no. 85, 1932. Washington, D. C.
Thompson, T. G., and R. J. Robinson. 1939.
“Notes on the determination of dissolved oxygen in sea water”
. Jour. Marine Research, v. 2, p. 1–8, 1939.
Thompson, T. G., and H. J. Taylor. 1933.
“Determination and occurrence of fluorides in sea water”
. Ind. Eng. Chem., Anal. ed., v. 5, p. 87–89, 1933.
Thompson, T. G., and T. L. Wilson. 1935.
“The occurrence and determination of manganese in sea water”
. Amer. Chem, Soc., Jour., v. 57, p. 233–36, 1935.
Thompson, T. G., and C. C. Wright. 1930.
“Ionic ratios of the waters of the North Pacific Ocean”
. Amer. Chem. Soc., Jour., v. 52, p. 915–21, 1930.
Tourky, A. R., and D. H. Bangham. 1936.
“Colloidal silica in natural waters and the “silicomolybdate” colour test”
. Nature, v. 138, p. 587–88, 1936.
Wattenberg, H.1933.
“Über die Titrationsalkalinität und den Kalziumkar-bonatgehalt des Meerwassers. Deutsche Atlantische Exped”
. Meteor 1925–1927, Wiss. Erg., Bd. 8, 2 Teil, pp. 122–231, 1933.
Wattenberg, H.1936. Kohlensäure und Kalziumkarbonat im Meere. Fortschritte d. Mineral., Kristal. u. Petrographie, v. 20, p. 168–95, 1936.
Wattenberg, H.1937. Critical review of the methods used for determining nutrient salts and related constituents in salt water. 1. Methoden zur Bestimmung von Phosphat, Silikat, Nitrat und Ammoniak im Seewasser. Conseil Perm. Internat. p. l'Explor. de la Mer, Rapp. et Proc.-Verb., v. 103, pt. 1, p. 1–26, 1937.
Wattenberg, H.1938. Zur Chemie des Meerwassers: Über die in Spuren vorkommenden Elemente. Zeitschr. f. anorg. u. allgemeine Chemie, v. 236, p. 339–60, 1938.
Wattenberg, H., and E. Timmermann. 1936. Über die Sättigung des Seewassers an CaCO3, und die anorganogene Bildung von Kalksedimenten. Ann. d. Hydrogr. u. Mar. Meteor., p. 23–31, 1936.
Wattenberg, H., and E. Timmermann. 1938. Die Löslichkeit von Magnesiumkarbonat und Strontium-karbonat in Seewasser. Kieler Meeresforschungen, Bd. 2, p. 81–94, 1938.
Webb, D. A.1937.
“Studies on the ultimate composition of biological material. Pt. 2. Spectrographic analyses of marine invertebrates with special reference to the chemical composition of their environment”
. Roy. Dublin Soc., Sci., Proc., v. 21, p. 505–39, 1937.
Webb, D. A.1938.
“Strontium in sea water and its effect on calcium determinations”
, Nature, v. 142, p. 751–52, 1938.
Webb, D. A.1939.
“The sodium and potassium content of sea water”
. Jour. Exper. Biol., v. 16, p. 178–83, 1939.
Wiese, W.1930. Zur Kenntnis der Salze des Meereises. Ann. d. Hydrogr. u. Mar. Meteor., Jahrg. 58, p. 282–286, 1930.