Chapter Eleven
Congenital Heart Disease
Many imperfections may be found in the heart of a newborn. In approximately 1 percent of live births there is a significant malformation of the heart that may affect the child's health. Such congenital defects may be restricted to the heart or may also show up in other organs and parts of the body. These malformations fall into three groups:
defects having no effect on the function of the heart or on the development of the child but sometimes leading to problems later in life (such as a bicuspid aortic valve)
defects affecting the function of the heart but compatible with normal or near-normal development and survival to adulthood
defects incompatible with survival beyond infancy
In some cases the cause of the defect can be identified. Genetic and environmental factors may be responsible for malformations of a fetus. Genetic errors involving identifiable chromosomal abnormalities usually produce multiorgan syndromes (the best known is Down's syndrome), which frequently involve the heart; genetic errors affecting the heart alone sometimes appear in several members of a family. However, genetic factors account for only about 8 percent of cases of congenital malformation of the heart. Certain environmental agents cause congenital cardiac malformations if the mother is exposed to them during the early stages of pregnancy. These include viral infections (particularly maternal German measles),
drugs, tobacco, and alcohol. Malnutrition of the mother can also lead to such defects.
The effect of cardiac birth defects on the function of the heart and circulation is related to two basic factors—stenosis of various points of the circulation (usually cardiac orifices or the aorta) and abnormal communication between the two sides of the circulation. Abnormal openings or ducts connecting the arterial and venous sides of the circulation are unique to congenital heart disease, and their size and location usually determine the seriousness of the malformation.
The normal pathway of fetal circulation is illustrated in figure 34. The principal difference between the fetal circulation and the circulation after birth is that in the former the process of acquiring oxygen and disposing of carbon dioxide takes place in the mother's placenta rather than in the lungs. During the second and third trimesters of pregnancy all the tissues of the body are formed and require a steady supply of oxygen. Oxygenated blood from the mother's placenta enters the fetal inferior vena cava through the umbilical vein and hence into the right side of the heart. From there part of the blood enters the left atrium through the foramen ovale , and the rest is propelled into the pulmonary artery. But instead of flowing into the inactive lungs, most of this remainder is directed into the descending aorta through a special duct, the ductus arteriosus . Whereas in the permanent postnatal circulation fully oxygenated "pink" blood fills the systemic arterial system and the deoxygenated "blue" blood the systemic venous system (each totally separate), the fetus lives on a mixture of oxygenated and deoxygenated blood. Oxygenated blood from the placenta and deoxygenated blood returning from the fetus's body are mixed in the right atrium and shunted into the left side of the heart, supplying the head and upper extremities. Most of the blood ejected into the pulmonary artery is deflected through the ductus arteriosus into the descending aorta, supplying the lower part of the body. The tissues of the fetus adapt to the lower oxygen content of this blood, although it gives the outward appearance of the fetus a purplish tinge.
After birth the two fetal structures permitting communication between the two sides of the circulation—the foramen ovale and the ductus arteriosus—close, but in some cases they may remain

Figure 34. The fetal circulation.
open, providing an abnormal pathway for blood mixing. In addition, other abnormal communications may permit blood mixing as a result of fetal malformation.
The mixing of fully oxygenated arterial blood and deoxygenated venous blood is a unique feature of some congenital cardiac malformations. When the admixture of venous blood in the arteries reaches a certain level it results in cyanosis , a purplish or bluish
discoloration of the skin and lips. Temporary cyanosis, particularly of the extremities, is a common phenomenon in healthy subjects, for whenever the circulation through a part of the body slows down (for example, because of exposure to cold or the placement of a tourniquet), more oxygen is extracted from the blood by the tissues. Slowed circulation in patients who are in heart failure may produce cyanosis of the entire body. Under these conditions the arterial blood is fully oxygenated, but the oxygen content of the venous blood is unusually low (peripheral cyanosis ). In central cyanosis , however, it is the arterial blood that contains a reduced amount of oxygen; its most common cause is blood mixing. Another mechanism of central cyanosis is deficient oxygen exchange in the lungs in some pulmonary diseases.
Blood mixing in congenital heart disease occurs in one of two ways: as a shunt, when a certain volume of deoxygenated blood enters the left side of the heart or the aorta, or as complete mixing (analogous to fetal circulation), when the arterial and venous blood mix freely in some part of the heart. Either mechanism results in reduced oxygen saturation of the arterial blood. Normal oxygen saturation is at least 95 percent; small or moderate shunts may reduce oxygen saturation to 85–90 percent. The normal extraction of oxygen by tissues remains unchanged when arterial oxygen saturation is reduced; hence oxygen saturation of the venous blood drops. Large shunts or free mixing of blood may reduce arterial oxygen saturation to as low as 60 percent. Cyanosis in infants, colloquially termed "blue baby," becomes visible at an oxygen saturation level of about 85 percent, though the threshold can vary widely.
Cardiovascular shunts allow the flow of blood through an abnormal opening or duct between the arterial and venous circulations. Intracardiac shunts result from defects in the atrial or ventricular septum; a common extracardiac shunt is due to a persistent fetal ductus arteriosus. In the postnatal circulation the pressure in the left heart chambers or the aorta is higher than in the corresponding parts of the right heart and pulmonary artery; hence the usual direction of the shunts is into the right side of the circulation (left-to-right shunt ). When the fully oxygenated blood in the left heart and aorta reenters the right side, there is no effect on oxygen saturation of either arterial or venous blood. Under abnormal circumstances
shunting may occur in the opposite direction (right-to-left shunt ), in which case lower oxygen saturation and cyanosis develop.
Narrowing of the cardiac orifices may involve the valves (congenital valvular stenosis ) or other structures where blood flows into and out of the heart. A relatively common malformation is stenosis of the aorta at a point where the aortic arch joins the descending aorta (coarctation of the aorta ).
The complexity of congenital malformations of the heart, particularly in infants, presents a challenge in establishing the correct diagnosis. Some of the simpler defects can be recognized in infancy by the presence of heart murmurs. Intracardiac communications may cause the blood flow through the abnormal defect to be turbulent, thereby producing a murmur. Murmurs of ventricular septal defect and patent ductus arteriosus are sufficiently characteristic to suggest the diagnosis within the first year of life. Noninvasive cardiac tests provide further clues to the diagnosis. Developments in echocardiography have revolutionized the diagnosis of congenital heart disease, permitting a definitive diagnosis of some of the more complex malformations without invasive testing. However, in many cases it is still necessary to resort to a combination of cardiac catheterization and angiocardiography.
Fetuses can be afflicted with a great variety of cardiac malformations, some of which cause stillbirth or death immediately after birth. Survivors' prognosis ranges from death in infancy to normal development and normal life span. The following discussion will present in detail six congenital lesions that account for more than 90 percent of congenital heart diseases permitting survival to adulthood (even without surgery).
Atrial Septal Defect
During fetal life communication between the two atria is essential for the fetus's survival. The foramen ovale, through which blood enters the left atrium, is protected by a valve that permits flow only from right to left. At birth, when pressure in the left side of the heart rises after the newborn takes its first breath, the foramen ovale ceases to function. Ordinarily it seals itself off, but even in cases when the sealing fails to occur, the valve does not permit it to function
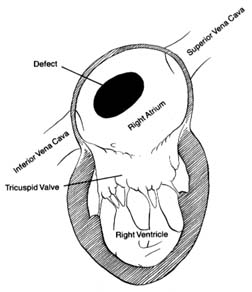
Figure 35. A typical atrial septal defect
as a pathway for blood flow. Atrial septal defect is an abnormal opening, usually located in the region of the foramen ovale, but larger and wide-open, allowing large quantities of blood to flow from the left to the right atrium (fig. 35). The quantity of blood shunted through this communication varies according to the size of the defect and the capability of the right ventricle to accept the additional blood. Small atrial shunts have no serious effect on the circulation since left-to-right shunts do not reduce the oxygen content of the blood. Large shunts, however, can overload the right side of the heart and increase blood flow through the lungs as much as fourfold. Normally each heartbeat ejects from each ventricle into the respective arteries (aorta and pulmonary artery) an average of 75 cc of blood. In the case of a large atrial septal defect right ventricular ejection may increase to 300 cc, and left ventricular output remains normal; hence 225 cc of blood returns to the right side of the heart during systole. This is referred to as a four-to-one shunt (a ratio of pulmonary to systemic output). A large shunt not only overloads the right ventricle, producing its hypertrophy and dilatation, but the wear and tear on the small arteries in the lungs may damage their inner layer, which in some persons can lead to high pressure in the
pulmonary arterial circulation. Pulmonary hypertension develops in about 15–20 percent of patients with atrial septal defect, almost always as adults. It may progress to the point where pressure in the right side of the heart exceeds that in the left, reversing the direction of blood flow from right to left through the shunt.
An atrial septal defect generally goes undetected until late childhood, adolescence, or even adulthood. The reason is that the flow through the defect between two low-pressure chambers does not produce turbulence audible as a murmur on physical examination. The child grows and develops normally (no cyanosis is present), except that the increased blood flow through the lungs raises susceptibility to respiratory infections. The diagnosis may be suggested on physical examination in older children by certain subtle abnormalities, but it is usually made when a chest X ray is taken, showing an enlarged heart and full blood vessels in the lungs. Confirmation of the diagnosis comes from echocardiography and cardiac catheterization, which reveals the magnitude of the shunt and the presence or absence of pulmonary vascular reaction (hypertension).
The course of atrial septal defect usually creates only minor problems. Development of the affected child is usually normal, although sometimes growth may be slightly impaired. Tolerance for exercise may be below average but seldom to the point of suggesting some disability. Frequent respiratory infections may occur. Persons in whom the defect is not surgically corrected may not develop significant symptoms until the fourth or fifth decade of life—sometimes even later. Reactive pulmonary hypertension, affecting one out of five patients with atrial septal defect, usually appears in early adulthood.
The treatment for atrial septal defect is surgical closure. This is one of the simplest cardiac operations, but it does require a pump-oxygenator. The operation is usually performed in older children or adolescents. Because of the risk of pulmonary hypertension it is best to close the defect before the age of 20. Closure of atrial septal defect is almost always a prophylactic operation performed on asymptomatic patients. If pulmonary hypertension is already present, closure may no longer be effective in preventing its progression. If cyanosis develops, closure of the defect is contraindicated since pulmonary hypertension then becomes the principal abnormality, which the operation will not correct.
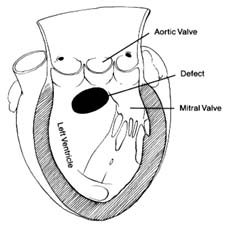
Figure 36. A typical ventricular septal defect.
Ventricular Septal Defect
Defect in the septum between the two ventricles is analogous to atrial septal defect in that it causes a unidirectional shunting of blood (fig. 36). An important difference between the two, however, makes ventricular septal defect a rather unique cardiac lesion. The two circulations, systemic and pulmonary, are closed systems: the former operates under high pressure determined by the resistance of small arteries, the latter under low pressure related to the minimal resistance of small branches of the pulmonary artery. Communication between the two circulations through the atria, where pressure in both chambers is close to zero, permits shunting of blood from one side to the other without significant consequences other than overload. However, communication between them in areas of high pressure—the ventricles or the arteries—may have disastrous consequences. A large opening between the two ventricles, with a systolic pressure of 120 mm Hg on the left side and 20 mm Hg on the right side produces a pressure gradient, so that most of the blood might escape to the right ventricle instead of entering the aorta. Clearly, a person could not survive were this allowed to happen; hence in compensation the systolic pressure in the right ventricle rises to match that in the left. Right ventricular systolic pressure can be elevated by one of two mechanisms—abnormal constriction of the small pulmonary arteries, increasing resistance
to a level comparable to that of the systemic arteries and thereby producing pulmonary hypertension; or pulmonary valve stenosis.
A small ventricular septal defect is characterized by a left-to-right shunt through the opening because it offers enough resistance to blood flow to maintain the normal difference in pressures between the two ventricles. The magnitude of the shunt entirely depends on the size of the opening, which ranges from that of a pinhead to that of a dime. For the average adult heart the largest diameter consistent with a normal pressure relationship between the two ventricles is about 1.5 cm, the approximate dividing line between small and large ventricular septal defects. The blood flow through a ventricular septal defect produces turbulence and generates loud heart murmurs audible on examination of a child; hence the diagnosis of such a defect is usually made at birth or at least during infancy. The hemodynamic consequence of a small ventricular septal defect varies with the magnitude of the shunt. Small defects shunting no more than half of the left ventricular output into the right ventricle have no effect on circulatory dynamics. Although the heart murmur is still detectable, no other abnormalities are usually discovered. The prognosis is favorable, with a normal life expectancy. The only risk is the possibility of infective endocarditis. Surgical correction is usually considered unnecessary. A significant number of such patients experience spontaneous closure of the defect—in essence, self-cure. Larger shunts through small ventricular septal defects (greater than twice the left ventricular output) have effects similar to those of atrial septal defect, namely overload of the heart and overfilling of the pulmonary blood vessels. Since ventricular septal defect with large pulmonary flow but normal right ventricular pressure takes a similar course as atrial septal defect, surgical closure (by means of open-heart surgery) is generally performed in early childhood.
Large ventricular septal defect is a more serious problem. The normal function of the left ventricular pump has to be protected by pressure elevation in the right ventricle from birth; hence pulmonary hypertension is already present in infancy. The left-to-right shunting of blood is usually considerable. Persistent high pressure in the pulmonary arterial system, together with large blood flow in the pulmonary circulation, may gradually increase the resistance to the point of reversing the shunt from right to left. This complication
happens occasionally in late childhood but most frequently in adulthood. Large ventricular septal defect complicated by shunt reversal and resulting in cyanosis is called Eisenmenger's syndrome . Despite pulmonary hypertension the growth and development of children with large ventricular septal defect are usually normal, and disability is rare. Even a fully developed cyanotic stage due to shunt reversal may be well tolerated, although in that case life expectancy is greatly reduced.
The treatment for large ventricular septal defect is surgical closure. The goal is to prevent progressive disease of small pulmonary arteries; thus surgical closure is often performed on small children, even infants. In the case of a large left-to-right shunt, closure of the defect may significantly reduce the pressure in the right ventricle and lower or even eliminate pulmonary hypertension. Large ventricular septal defect also occurs in combination with pulmonary stenosis, in which case pulmonary hypertension is absent (see discussion of tetralogy of Fallot below).
Patent Ductus Arteriosus
During the fetal period the ductus arteriosus connecting the descending aorta with the pulmonary artery serves as an essential pathway supplying blood partly oxygenated by the placenta to the arteries of the lower part of the fetal body. Normally the duct closes immediately after birth, but owing to a developmental error it may remain open, or patent, in some infants. The altered pressure conditions during postnatal life (higher in the systemic circulation than in the pulmonary circulation) allows blood to flow through the duct in the opposite direction, that is, from the aorta to the pulmonary artery. This left-to-right shunt on the arterial level is analogous to the atrial and ventricular septal defects. As in the other two conditions, fully oxygenated blood returns to the lungs; gas exchange proceeds normally, but the excess blood may overfill the pulmonary circulation. Since that excess has to be pumped into the aorta by the left ventricle, the latter may be affected by the increased workload if the shunt volume is large.
The size of persistent ducts varies widely. A small duct has no significant effect on circulatory dynamics but is at risk of developing infective endarteritis (an infection analogous to endocarditis,
striking the arteries rather than the heart). The turbulent flow through the duct produces an audible murmur in the patient's chest different from murmurs caused by lesions inside the heart. A large duct, by overloading the left ventricle and the pulmonary circulation, may produce pulmonary hypertension in adulthood. Rarely, a very large duct shunts so much blood away from the aorta of an infant that a surgical emergency is created.
Surgical correction of patent ductus arteriosus consists of tying off the duct in an operation that does not require use of the pump-oxygenator. The duct can also sometimes be closed through a non-surgical technique involving cardiac catheterization. The simplicity and low risk of duct closure justifies its performance even in cases where the condition has no effect on the circulation, to guard against infection. Spontaneous closure of the duct occurs in infancy or childhood in an estimated 5 percent of cases.
Pulmonary Stenosis
Congenital stenosis of the outflow segment of the right ventricle may be a sole malformation or part of a complex combination of defects. The simplest variety of pulmonary stenosis is fusion of the three leaflets of the pulmonary valve into a single membranelike structure with an opening in the center. As in other forms of valvular stenosis, the size of the opening directly determines its effect on circulatory dynamics. Pulmonary stenosis, if significant, places a pressure overload on the right ventricle analogous to the effect of aortic stenosis on the left ventricle (see chap. 9). The resistance to outflow from the right ventricle elevates the pressure inside that chamber, causing a pressure gradient between the ventricle and the pulmonary artery, where the pressure remains normal. In health identical pressures in the right ventricle and the pulmonary artery in systole average 15 mm Hg. In mild pulmonary stenosis right ventricular systolic pressure may rise to about 40 mm Hg; in moderate, to 70 mm Hg; and in severe, to 150 mm Hg. The right ventricle is quite able to adapt to the high pressure, particularly in infancy. However, in severe pulmonary stenosis the overloaded right ventricle may eventually produce right ventricular failure. In rate cases severe pulmonary stenosis may lead to serious emergencies in infancy requiring immediate surgical intervention.
Mild cases of pulmonary stenosis require no treatment other than preventive measures against endocarditis. Severe pulmonary stenosis requires surgical relief. Pulmonary valvotomy by means of open-heart surgery was the standard treatment until the late 1980s. Balloon dilatation of the pulmonary valve has now been applied successfully and is becoming the treatment of choice. The use of prosthetic valves in pulmonary stenosis is rarely necessary.
Tetralogy of Fallot
Tetralogy of Fallot represents a combination of pulmonary stenosis and large ventricular septal defect. It is the most important malformation associated with persistent cyanosis, the commonest cause of "blue baby." The term "tetralogy" was suggested more than a century ago, when two other features (making a total of four) were considered central to this malformation. These two features are now known to be consequences of the first two.
The most essential component of this lesion is large ventricular septal defect. The obligatory high pressure in the right ventricle is, however, maintained by pulmonary stenosis rather than by high resistance in the pulmonary circulation, as is the case in isolated ventricular septal defect. The narrowing may be located either at the valve or below the valve (subvalvular pulmonary stenosis ). In some cases not only is the valve or the outflow segment of the right ventricle affected by the malformation, but the pulmonary artery may be smaller than average (hypoplastic ), compounding the difficulty in the pulmonary circulation. In most cases the resistance to outflow from the right ventricle is higher than that from the left ventricle—hence the reversal of blood flow (right-to-left shunt) resulting in cyanosis. When pulmonary stenosis is relatively mild, the usual left-to-right shunt through the ventricular septal defect takes place (noncyanotic tetralogy of Fallot). The wide variation in the resistances within the pulmonary outflow tract creates a range of skin coloration in children with tetralogy of Fallot—some showing normal color, others a faintly blue tinge, still others a deep purple color. In black children and other children with naturally dark skin, the effects of cyanosis may be less apparent. Despite the complexity of this malformation of the heart, survival beyond infancy is possible in most cases; development may be mildly impaired,
but many patients reach adulthood, some even surviving beyond middle age.
One of the most spectacular advances in treating congenital heart disease was the operation introduced in 1944 by Alfred Blalock and Helen Taussig. This procedure crafted a connection between the systemic circulation and the pulmonary circulation by suturing the large artery supplying the arm (brachial artery) into the pulmonary artery branch; the arterial blood, poorly oxygenated because of mixing with venous blood through the right-to-left shunt, could thereby return to the lungs for reoxygenation. The drama of deeply blue children assuming a pink color after the operation created a sensation when it was first performed. This operation is now rarely used since it does not attack the basic defect. Open-heart surgery allows surgeons to close the ventricular septal defect and dilate the right ventricular outflow tract; the procedure is performed on patients in infancy or early childhood.
Coarctation of the Aorta
It is customary to include in any discussion of congenital heart disease coarctation of the aorta , even though it is a vascular, not a cardiac, malformation. It involves a congenital narrowing of the aorta in the region where the aortic arch changes into the descending aorta. This stenosis is usually severe, bordering on complete interruption of flow into the descending aorta. The flow of arterial blood into the head and upper extremities is unimpaired, but the rest of the body has to receive blood via a detour—collateral circulation. Branches of the aorta supplying blood to the upper part of the body form connections with branches of the descending aorta, especially arteries supplying the chest that run between the ribs. These connections function so that when some of the arterial blood meant for the upper parts of the body reaches the arteries in the chest, it flows in the opposite direction, into the descending aorta. As a consequence of the abnormally high resistance caused by redirection of the blood, patients usually suffer from high blood pressure, which may overload the left ventricle and cause its hypertrophy.
Coarctation of the aorta accounts for many cases of high blood pressure in infants, children, and adolescents. Such hypertension is curable, disappearing when the coarctation is corrected. Although
hypertension is the most serious consequence of coarctation, rare complications include infective endarteritis, stroke caused by small aneurysms of an artery supplying the brain, and aortic dissection (see chap. 14).
Other Congenital
Malformations of the Heart
Transposition of the great arteries is a complex but common malformation. Before surgical procedures allowing partial or total correction were developed, few afflicted with this defect survived childhood. With surgical treatment patients may now reach adulthood, though their life expectancy remains greatly reduced. This malformation consists of an error in cardiac development wherein the aorta arises from the right ventricle and the pulmonary artery from the left (fig. 37). The transposition creates two separate closed circulations incapable of the exchange of gases in the blood necessary for survival: deoxygenated blood returning to the right heart through systemic veins is ejected into the aorta without being able to acquire oxygen and dispose of carbon dioxide. Blood returning fully oxygenated from the lungs enters the pulmonary artery and returns to the lungs. Obviously, this situation is incompatible with survival unless some mixing of the two bloods takes place. That need is met by persistence in the infant of the fetal communication between the two circulations, namely the foramen ovale. Occasionally ventricular septal defect is also present and assists in the mixing. Infants surviving on this oxygen-poor blood have a dark purple coloration of the skin; if a sizable proportion of the oxygenated blood is shunted into the arterial side, survival beyond infancy is possible without operation. Several methods of correcting the defect are now available, restoring normal color and permitting survival into adulthood.
Pulmonary atresia consists of total closure of the outlet for blood from the right ventricle. All the venous blood returning to the right side of the heart is shunted to the left side through atrial and sometimes ventricular septal defects. The fully mixed blood reaches the pulmonary artery and the lungs via a patent ductus arteriosus. Deep cyanosis is usually present.
Tricuspid atresia involves an absence of communication between
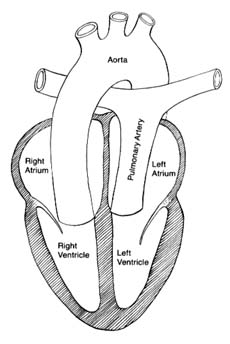
Figure 37. The circulation in complete transposition of the great vessels.
Communication between the two sides of the heart, necessary
for survival, is not shown in the drawing.
the right atrium and the right ventricle. Venous blood is shunted through a defect into the left atrium, and the mixed blood enters the lungs through the ductus arteriosus.
Total anomalous venous return consists of malposition of the pulmonary veins carrying oxygenated blood, which enter the right atrium instead of the left atrium, resulting in total mixing of unoxygenated and oxygenated blood. The mixture is shunted through an atrial septal defect into the left atrium and is ejected by the left ventricle in the aorta.
Ebstein's anomaly involves malposition of the tricuspid valve well inside the right ventricle, reducing its capacity. The foramen ovale often remains patent, allowing a portion of the blood from the right atrium to be shunted to the left side. The extent of cyanosis varies according to the size of the right-to-left shunt through the
foramen ovale. This lesion permits survival to adulthood and occasionally to old age.
Truncus arteriosus is a malformation of the great arteries attached to the heart. A single artery is connected with both ventricles, which communicate through a ventricular septal defect. This arterial trunk divides into a pulmonary artery and aorta. Total mixing of the blood occurs in the trunk.
Single ventricle occurs when the entire septum between the two ventricles is absent. This malformation produces complete mixing of oxygenated and unoxygenated blood.
These seven malformations have in common the mixing of oxygenated and unoxygenated blood in the systemic arteries. As a consequence, the tissues receive blood poorer in oxygen than is normal. Survivors are capable of growth, development, and some activity. Surgical treatment is now available for most of these syndromes, with variable results.
Congenital malformations without shunting of blood involve abnormalities of cardiac valves. Pulmonary stenosis has already been discussed in this chapter; left-sided cardiac valvular diseases are presented in chapter 9. Aortic stenosis is a relatively common congenital malformation, mostly as a valvular stenosis involving fusion of the three leaflets of the aortic valve. Rare variants of stenosis of the left-sided outflow segment of the ventricle include subvalvular stenosis , in which a membrane with a central opening stretches across the outflow segment underneath a normal aortic valve, and supravalvular stenosis , a narrowing of the aorta just above the aortic valve. A common anomaly of the aortic valve is the bicuspid valve (two leaflets instead of three), which has no effect on the heart and circulation but may be the site for deposition of calcium late in life and hence a cause of calcific aortic stenosis. Congenital mitral stenosis is rare as an isolated malformation, although it is seen in combination with other defects.