Williamson's Asymmetric Synthesis Argument
Williamson's work proved to be the pivot on which turned the ultimate breakthrough of the movement as a whole, because an effective experimental argument that he devised in 1850 and that saw various applications in the next five years essentially settled the molecular magnitude issue, provided compelling grounds for adopting the newer type theory and the atomic weight reform, and further weakened the vestiges of dualism enshrined in the copula theory. During his student years in the 1840s in Heidelberg, Giessen, and Paris, Williamson (1824-1904) was a student of Gmelin and Liebig, and he became the first major advocate of Laurent's and Gerhardt's chemistry.[5] In 1850, newly installed as professor of analytical chemistry at University College London, he decided to try to settle the important issue of relative molecular magnitudes of alcohol (in modern terms, CH3 CH2 OH or Et-OH) and ether (CH3 CH2 OCH2 CH3 or Et-O-Et).[6]
From the 1820s through the 1840s, Liebig and most other chemists had assumed that alcohol was simply the hydrate of ether, since acid dehydration of alcohol yields ether—this despite the fact that the vapor density of ether is nearly twice that of alcohol.
![]()
Laurent and Gerhardt, however, argued from 1846 on that alcohol is a monoethyl and ether is a diethyl water derivative. Reacting Frank-land's reagent ethyl iodide together with a substance first prepared by Liebig, potassium ethoxide, Williamson succeeded in substituting ethyl for the replaceable hydrogen of alcohol, resulting in a novel ether synthesis that was elegant, smooth, low-temperature, and high-yield. In Williamson's terms,
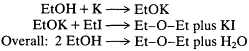
This reaction could still be explained by the older theory if one assumed that the potassium ethoxide and ethyl iodide were each separately transformed, yielding two ether molecules rather than one. But an additional advantage of the Williamson reaction was its flexibility in producing new "tailored" ethers, and an ingenious modification by Williamson cut off any retreat. By reacting methyl iodide with potassium ethoxide, the two theories would now predict two distinct results. The Gerhardt-Laurent (Williamson's) theory required the production of a single, homogeneous product, an asymmetric ethyl methyl ether. Liebig's theory would require a mixture of products to result from the reaction, the symmetrical (normal) ethyl ether, derived from the ethoxide, and a new symmetrical methyl ether, derived from the iodide.
![]()
The consequent production of a single ethyl methyl ether settled the issue, at least for Williamson. As Gerhardt and Laurent had been urging, the ether molecule must be essentially twice the size of the alcohol molecule, not simply a dehydrated version of it; more generally, there is no pre-formed water in alcohols or acids, nor pre-formed oxides in salts. The Gerhardt-Laurent molecular magnitudes reform appeared to be established at a stroke, and dualistic formulations were further weakened, if not destroyed. Many chemists agreed with Williamson, as we shall see in the next section.
The atomic weight reform required another kind of argument. The focus of the ether molecule was the oxygen, which Williamson saw no reason to subdivide. A single oxygen atom, O = 16, could then serve as the material link or bond between two ethyl radicals in ether, an ethyl and a hydrogen in alcohol, or two hydrogens in water. The argument
was cogent, but not as compelling as that for the structure of ether; it involved an appeal both to Ockham's razor and to structuralist instincts.[7]
The best evidence for the strong impact of Williamson's asymmetric synthesis argument is its widespread deployment in the five years after 1850. In 1851 Williamson used it again himself, in conjunction with a messy ketone reaction, to advocate Gerhardt's and Laurent's formula for acetone—and solving one of their troubling anomalies in the process. As we saw in chapter 4, in the spring of 1852 Gerhardt synthesized organic acid anhydrides by a reaction route analogous to Williamson's (and already predicted by Williamson the year before). He also applied Williamson's logic as well, by producing asymmetrical anhydrides (such as acetic-benzoic anhydride) that could be explained only by the new chemistry. Gerhardt thought this accomplishment was "terribly revolutionary," the best paper of his life. Indeed, it convinced a wide variety of skeptics both that monobasic organic acids contain no water even in their "hydrated" form and that the water molecule must have two hydrogen atoms. This was the event that propelled Gerhardt from his pariah status to career success—if unfortunately only briefly, as he died in 1856.
The next chapter will show how in 1854 William Odling and August Kekulé, then living in England, independently applied versions of Williamson's argument. Also in 1854 Williamson himself used it once more. He succeeded in chlorinating sulfuric acid in two separate stages, producing in the first reaction a substance now known as chlorosulfonic acid, HO.SO2 .Cl. If one were to reason on the basis of the conventional formula for sulfuric acid, HO.SO3 , the halfway chlorination ought to have produced at best a mixture of HO.SO3 and Cl.SO3 , and not a homogeneous asymmetric product. It was clear to Williamson from this reaction that there is no preformed water even in dibasic inorganic acids. Again, the reaction had indicated the inconsistent molecular magnitudes of the hitherto prevailing views: the conventional sulfuric acid formula HO.SO3 , like the HO formula for water, needed to be doubled to compare consistently with the usual organic formulas (in more unitary, nondualistic terms, H2 S2 O8 ), and then the larger Gerhardtian atomic weights had to be applied to oxygen and sulfur (yielding H2 SO4 ). Viewed in this light, his argument was that, in sulfuric acid, the sulfuryl radical SO2 is the material link between HO and Cl in chlorosulfonic acid and between two HOs in sulfuric acid, just as oxygen is the linking element in ether and in water.[8]
Assumptions of relative molecular sizes and atomic weights were also crucial in interpreting the new radicals created by Kolbe and Frankland during the years from 1847 to 1850. The radicals formed in
these reactions are now thought to combine together as they are formed, to create dimers:
![]()
Nothing in their reactions of formation, however, indicates this dimerization, and Kolbe and Frankland wrote versions of the simpler equations as follows:
![]()
These implied production of the actual monomeric radicals. Although Gerhardt and Laurent immediately argued for the dimer formulas of the new substances, it is understandable that Kolbe and Frankland, and initially most other chemists, were not persuaded of the truth of a more complex and less obvious reaction route—especially because the central argument that the Frenchmen used was their mysterious and unmotivated even-number rule.[9]
But by March 1850, newly converted from copula to type formulations, Hofmann became convinced that Gerhardt and Laurent were right.[10] He attempted to settle the issue by an experiment designed to decarboxylate valeric acid pyrolytically, but the results of the experiment were messy and unhelpful. Still, he appended a thorough brief for the thesis that the "radicals" were dimeric molecules. They had none of the properties that one would expect from true radicals, such as extreme reactivity, or additive reactions with oxygen or halogens. That they had proven so difficult to extract from their compounds suggested all the more strongly that they ought, if they were true monomeric radicals, to have properties similar to the highly active "radicals" (i.e., metals) extracted by Davy from soda and potash. On the contrary, the new hydrocarbons had all of the indifferent paraffinic properties of the predicted homologues of marsh gas.
Another anomaly was that the differences between the boiling points of three adjacent homologous "radicals" were found in each case to be 47ºC. This was about twice the fairly consistent boiling point differences between nearly all adjacent homologs, as found by Hermann Kopp. The anomaly would vanish if the formulas for the "radicals" were doubled and intermediate unknown hydrocarbons were interpolated. Again, whereas amylene (C10 H10 ) and amyl hydride (C10 H12 ) boil at 39ºC and 30ºC, respectively, "amyl" (Frankland's and Kolbe's C10 H11 ) boils at 155ºC, which did not seem reasonable to
Hofmann; a doubled formula would again restore consistency in the place of anomaly. Finally, the "radicals" all had doubled vapor densities compared to similar olefins and hydrides.
In short, both physical and chemical properties suggested that these molecules were twice the size of what Frankland and Kolbe thought. Hofmann conceded that this meant supposing an in situ and in statu nascenti dimerization reaction, for which there was no direct evidence. He pointed out, however, that in situ polymerization or condensation reactions were already known and accepted, such as the formation of acetone from acetates and mesitylene from acetone.
In December 1850 Benjamin C. Brodie, Jr., adduced additional arguments in favor of the dimer formulas.[11] At the end of his paper, he suggested a crucial experiment modeled on Williamson's recently published asymmetric synthesis argument. Frankland had reacted ethyl iodide with zinc to produce what he thought was ethyl and what Brodie and Hofmann were interpreting as ethyl-ethyl. If one instead used mixtures such as ethyl iodide and amyl iodide for the reaction, one might be able to produce asymmetric radicals, in this instance ethyl-amyl. The formation of such an asymmetric radical as a product would demonstrate that dimerization was occurring, since according to Frank-land's conception one would expect to produce only mixtures of symmetrical radicals, ethyl and amyl. But Brodie reported that neither he nor Hofmann had had success with the reaction.
That success was achieved by Wurtz in 1855.[12] In place of Frank-land's, Brodie's, and Hofmann's zinc (Frankland, presumably following an earlier suggestion of Liebig's, had also tried potassium), Wurtz got the reaction to work using sodium, which he said combined the advantages of acting less energetically and being cheaper than potassium. Following the Williamsonian strategy indicated by Brodie and Hofmann,[13] Wurtz produced the "mixed" radicals ethyl-butyl, ethyl-amyl, buty-amyl, butyl-caproyl, and methyl-caproyl. The argument from this evidence for the larger dimeric formulas for the Kolbe-Frankland "radicals," averred Wurtz, was "of the same order and just as conclusive" as Williamson's for the larger ether formula. One of Hofmann's predicted interpolated compounds was also a product of this research, namely, butyl-amyl (in Wurtz' four-volume formulas, C8 H9 .C10 H11 ), which was half-way between "butyl" (Kolbe's "valyl," in Wurtz' formulation C8 H9 .C8 H9 ) and "amyl" (C10 H11 .C10 H11 ). The boiling point, Wurtz emphasized, just matched Hofmann's prediction. Wurtz even succeeded in producing similar asymmetric radicals from Kolbe electrolyses.
Dimerization introduced a new complexity into the explanation of the reactions, but Wurtz, again following Brodie and Hofmann, cited
other accepted instances of this kind of reaction. He was even able to make reference to a project he had carried out under Liebig in 1842 involving the reaction of hydrochloric acid and copper hydride with release of hydrogen gas, which supported Dumas' 1828 speculation on the "dimerization" of hydrogen atoms to form hydrogen molecules.[14] Wurtz also pointed out that this was one of the theses defended by Laurent when, in 1846, he clarified Gerhardt's ideas on atoms and molecules. He mentioned that it had also been proposed and defended more than forty years earlier by Ampère.
The "Wurtz reaction" is still taught in organic chemistry classes today, but it is a messy reaction producing problematical product mixtures, and it is no longer synthetically useful. This is all the more reason for admiring Wurtz' achievement in unraveling the complex chemistry and properly analyzing the liquid and gaseous products, then drawing the important theoretical conclusions—even if the latter were not original to him. He concluded the paper with an advocacy brief for the new chemistry. He used the new atomic weights in this section, "for greater simplicity,"[15] but in general continued to employ the older conventional equivalents for another four years.
It is interesting that all of the protagonists in the reform movement sketched here expressed themselves after the new type theory emerged in terms of compatibility and complementarity rather than conflict. In 1850, Frankland, Hofmann, Wurtz, and Williamson all implied independently that their work on organometallics, complex amines, primary amines, and ether, respectively, suggested a consolidation of radical and substitution theories.[16] The same year Gerhardt used similar language in attempting to reconcile with Liebig, and Liebig returned the rhetoric two years later.[17] Also in 1852, Frankland stated unequivocally that his and others' recent research promised "to assist in effecting a fusion of the two theories which have so long divided the opinions of chemists, and which have too hastily been considered irreconcilable."[18]
Finally, in 1855 Wurtz put forth an extended argument having to do with "condensed" or multiple types. He pointed out that although the old radical theory used a binary conception of constitution with addition mechanisms, the new chemistry used a similar binary constitutional orientation with substitution mechanisms. Thus, "far from being contradictory, they complement one another."[19] These visions of continuity and complementarity were clear-sighted and not simply an instance of soft-pedaling revolution. The newer type theory was a natural and logical outgrowth of the development of the science to 1849; it incorporated much of the older theories, while at the same time justifying many of the new ideas of Laurent and Gerhardt.