7
Government Inhibits Medical Device Distribution
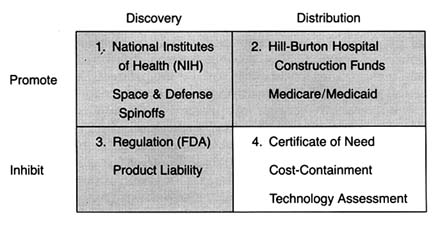
Figure 22. The policy matrix.
The federal and state payment programs described in chapter 4 provided little incentive for hospitals, providers, or eligible patients to consider costs when making decisions about health care. As a result, there were few economic barriers to the use of any new and apparently safe technology. Even the advent of regulation and the expansion of product liability in the 1970s did not appear to slow the growth of the health care technology market, at least in the aggregate.
However, concerns about the escalating costs of health care generated new attitudes toward medical delivery systems. The belief that more is better gave way to assertions that the system was too large, unwieldy, and wasteful. From the early 1970s through the mid-1980s, at least one dozen major federal laws
were enacted in response to spiraling medical costs.[1]
Lawrence D. Brown, "Introduction to a Decade of Transition," Journal of Health Politics, Policy and Law 11 (1986): 569-580, 571.
The dominant statutory and regulatory theme was cost containment. These new laws did not constitute revolutionary change. They did presage a conceptual shift, however, toward two potentially conflicting goals—promotion of competition in health care and greater government regulation and control.[2]Detailed discussion of these major and complex institutional reorganizations, such as HMOs (health maintenance organizations), capitation plans, and other forms of managed care, is beyond my scope here. For further reading, see Judith A. Hale and Mary M. Hunter, From HMO Movement to Managed Care Industry: The Future of HMOs in a Volatile Healthcare Market (Excelsior, Minn.: InterStudy Center for Managed Care Research, 1988); and Peter Boland, Making Managed Healthcare Work: A Practical Guide to Strategies and Solutions (New York: McGraw-Hill, 1991).
This chapter reviews the impact of government cost-control measures on the distribution of medical devices. Many considered medical technology a key culprit in the rising costs of health care. One widely cited 1979 study estimated that the contribution of new technology to hospital cost increases ran as high as 50 percent.[3]
Stuart H. Altman and Richard Blendon, Medical Technologies: The Culprit Behind Health Care Costs? Proceedings of the 1977 Sun Valley Forum on National Health (Washington, D.C., 1979), cited in Gloria Ruby, H. David Banta, and Anne K. Burns, "Medicare Coverage, Medicare Costs and Medical Technology," Journal of Health Politics, Policy and Law 10 (1985): 141-155, n.2.
There is no question that the cost-containment policies intended to slow the introduction and the diffusion of new technologies, particularly cost-raising products. The justification was that effective controls would not only reduce costs but also improve care because unconstrained diffusion led to excessive tests, treatments, and risks as well. (See figure 22.)There are three types of cost-containment strategies: behavioral, budgetary, and informational. Behavioral regulation tries to inhibit diffusion by modifying the behavior of medical decision makers. These policies influence decisions about use, expansion, and acquisition of technology. Prime examples of this policy strategy are the state based certificate-of-need programs (CON), described in more detail below. Budgetary regulation sets rates or expenditures, leaving administrators free to manage within the cost constraints established by the payers. Budgetary controls are epitomized by Medicare's prospective payment system (PPS). Informational regulation inhibits the adoption and diffusion of new medical technologies through prospective evaluation techniques, including technology assessment and the newer outcomes, or effectiveness, research.
Case studies illustrating the potentially powerful impact of these policies on the adoption and diffusion of medical devices include the intraocular lens (IOLs), artificial replacement lenses implanted after cataract surgery, and cochlear implants, permanently implanted devices that mitigate severe hearing impairment. Both of these cases demonstrate the impact of payment policies on the introduction of new devices. Also, they highlight the collective impact of a broad range of policies—from NIH, to
FDA, to HCFA—on the entire innovation process. By the 1970s and 1980s, policy proliferation was in full swing.
Policy Overview
Regulation of Market Behavior
The early cost-containment programs of the 1970s have been characterized as behavioral in that they tried to block decisions by providers about use, expansion, and acquisition of technology.[4]
See Brown, "Introduction to a Decade," in which he establishes these helpful categorizations of cost-containment policies.
The goal was to eliminate waste and unnecessary expenditure, in order to save the cost-based reimbursement system.One such effort was Title XVI of the National Health Planning and Resources Development Act of 1974, which supplanted the Hill-Burton program.[5]
David S. Salkever and Thomas W. Bice, Hospital Certificate-of-Need Controls: Impact on Investment, Costs, and Use (Washington, D.C.: American Enterprise Institute, 1979), 3.
These new controls stipulated that federal subsidies to eligible institutions must be made in compliance with statewide cost-control plans. In the 1970s, government subsidies became less important than other sources of funds, and the influence of these controls diminished considerably.In 1972, Congress also authorized establishment of federally funded, private Professional Standards Review Organizations (PSROs) to conduct independent quality and utilization reviews of hospital services under Medicare and Medicaid.[6]
Social Security Amendments of 1972, sec. 259F(b).
These efforts were highly controversial. Congress formally terminated the program ten years later, replacing it with an alternative peer-review approach. The Professional Review Organizations (PROs) established under the 1983 Medicare reform, discussed below, intended to monitor hospitals to determine if Medicare payments were appropriate.The statewide plans referred to in the health planning legislation generally meant certificate-of-need programs. New York state's passage of a certificate-of-need law in 1964 was the first government sponsored investment control. Concern about the inflation in health care costs led to the rapid diffusion of these types of controls. By 1978, thirty-eight states had adopted similar programs. State programs varied widely in terms of the types of institutions covered by the controls, thresholds for review, and legal sanctions for failure to comply.[7]
Clark C. Havighurst, "Regulation of Health Facilities and Services by Certificate-of-Need," Virginia Law Review 59 (October 1973): 1143-1232.
The goals of these laws generally were to eliminate "unnecessary" expansion of hospitals and to encourage less costly alternatives to hospital care. The easiest measure of these goals was excess bed capacity. Certificate-of-need agencies were predisposed to disapprove a hospital's proposal for new beds and spent considerable time and energy reviewing them. However, it is argued that controls were less successful when reviewing new services and equipment proposals, for several reasons: the costs of acquiring information about services and equipment are high, denials open the agency to charges of withholding from citizens the benefits of medical progress, and denials are likely to raise the ire of physician groups who want access to the same equipment that their colleagues have at other hospitals.[8]
Salkever and Bice, Hospital Certificate-of-Need, 20.
The evidence indicates that certificate-of-need programs resulted in no significant effect on total investments among hospitals but did alter the composition of those investments. Specifically, there was lower growth of bed supplies and higher growth of plant assets per bed. Thus, for most areas of medical device technology, these particular cost-containment strategies probably had little or no impact.Regulation through Budgetary Constraints
An alternative means to control hospital inputs is to put caps on reimbursements paid to hospitals from the federal and state governments. In September 1982, Congress directed the Department of Health and Human Services (HHS) to propose a plan to revise the Medicare payment system. That December, the Health Care Financing Administration (HCFA), the agency responsible for processing Medicare claims, proposed a prospective payment plan. Medicare had been building empirical data on this type of reimbursement scheme with demonstration projects in several states. Congress passed the Social Security Amendments of 1983 the following March, with most of the new provisions to be gradually phased in over a three-year period.[9]
Much has been written on the prospective payment system. See U.S. Congress, Office of Technology Assessment, Medicare's Prospective Payment System: Strategies for Evaluating Cost, Quality, and Medical Technology, OTA-H-262 (Washington, D.C.: GPO, October 1985). See also Louise B. Russell, Medicare's New Hospital Payment System: Is It Working? (Washington, D.C.: The Brookings Institution, 1989).
In brief, the plan created a complex prospective payment system, known as PPS. Medicare now bases its prices for Medicare hospital cases on a comprehensive classification system comprised of about 470 mutually exclusive categories called
"diagnostic related groups" (DRGs). The basic assumption is that all illnesses can be grouped according to disease system, intensity of resources consumed, and length of stay, among other categories, and that such groups reflect the average cost of providing services to all patients with diseases in that DRG. The price is then determined by calculating an average price per case for all Medicare cases plus the weight of the DRG assigned to the particular patient's case. The hospital is reimbursed at a price set in advance for each DRG rather than the actual cost of treatment. As of 1988, DRG reimbursement applied only to inpatient care. Physicians' services continued to be paid out on the old "reasonable cost" basis, but Congress passed legislation to extend a form of prospective payment to physicians in the 1990s.[10]
In 1986, Congress created the Physician Payment Review Commission (PPRC) to advise it on reforms of the methods used to pay physicians under Medicare. Its guiding principle has been that payment reform should provide equitable payment to doctors, protect beneficiaries, and slow the rate of increase of Medicare expenditures. As part of Omnibus Budget Reconciliation Act of 1989 (OBRA), Congress enacted comprehensive reform of Medicare physician payments. For a detailed discussion of Medicare physician payment issues, see Physician Payment Review Commission, Annual Report to Congress, 1990 (Washington, D.C.). The new fee schedule will not be in place until 1992 at the earliest. Its impact on medical device markets cannot be predicted at this time.
Congress understood that the prospective rate-setting process needed the flexibility to respond to advances in all health care technology. Congress created the Prospective Payment Assessment Commission (ProPAC) to participate in the process of updating the hospital payment rates in an independent and public fashion. ProPAC members are appointed by the Office of Technology Assessment (OTA), which is a congressional advisory body, and ProPAC's seventeen members must represent a wide range of constituencies. Its responsibilities under the law are to make annual recommendations to HHS on the appropriate percentage change in Medicare payments for hospital services and to make recommendations on necessary changes in the DRGs, including the establishment of new groups, modification of existing groups, and changing their relative weights when appropriate. HHS is not bound to adopt the recommendations of ProPAC and, to date, has not done so in many cases.[11]
The law requires that ProPAC submit annual reports to Congress that contain its recommendations on updating the Medicare prospective payments and modifying the diagnosis related group (DRG) classification and weighing factors. Public Law 98-21, sec. 1886(e)(4). See, for example, Prospective Payment Assessment Commission, Medicare Prospective Payment and the American Health Care System: Report to the Congress (April 1985) and annual reports thereafter.
The intended effect of this dramatically new payment structure was to encourage efficient delivery of health care services in the hospital sector, ultimately reducing federal Medicare expenditures. The intention to stabilize or reduce the market size as well as to inhibit unnecessary expenditures on underutilized equipment was an important goal. Consistent reductions in aggregate equipment expenditures have not, apparently, occurred, although sales in some SIC categories leveled off for a time because of uncertainty about the future.
The true impact of the new program on medical device producers
has been mitigated somewhat because it has not been fully implemented. At the end of the 1980s, one major issue relating to medical equipment had not been resolved. Congress deferred a decision on how the system should treat major capital expenditures by hospitals. Many medical devices, such as diagnostic equipment and monitoring and anesthesia products, fall into this category.
Under the cost-plus system of reimbursement before PPS, hospitals prepared capital-cost reports for Medicare. Medicare paid 100 percent of the reasonable costs of capital equipment (defined as land, buildings, and movable equipment) that were attributed to the care of Medicare patients in each hospital cost center. This procedure was known as the capital-cost pass-through , and it provided incentives for hospitals to expand and improve their capital base. The payment program, along with the growth of private insurance that provided stable cash flow, improved the borrowing power of hospitals and encouraged them to finance construction and equipment acquisition through debt. Hospital capital pass-throughs accounted for about 8 percent of the total Medicare spending in fiscal 1984 (with 14 percent of that attributable to depreciation of movable assets—that is, devices), a modest but important aspect of the financial picture for hospitals.[12]
Senate Committee on Finance, Hospital Capital Cost Reimbursement Under the Medicare Program, prepared for the Congressional Research Service of the Library of Congress by Julian Pettengill, 5 November 1985. See also Ross M. Mullner, "Trends in Hospital Capital and the Prospective Payment System: Issues and Implications," in Henry P. Brehm and Ross M. Mullner, eds., Health Care, Technology, and the Competitive Environment (New York: Praeger, 1989).
When PPS passed in 1983, Congress initially excluded capital-related costs from the prospective payment system, deferring a decision on the issue until October 1986. At the time, Congress sought more information because of the complexity of hospital capital spending and because of the variations in investment cycles that might give arbitrary advantages to hospitals with recently completed capital expansion if controls were imposed on a specific date.[13]
Brehm and Mullner, Health Care.
Congress proposed several capital-cost plans in 1985, but voted in 1986 and 1987 simply to reduce the percentage of capital-cost pass-through payments (phased in from a 3.5 percent reduction in 1987 to a 15 percent reduction in 1988), without tackling the more difficult task of folding capital costs into the prospective payment system. These congressional efforts prevented HCFA from imposing its own rules.[14]
52 Federal Register 33168-33199 (1 September 1987).
By the end of the 1980s, then, there was a two-tiered payment
system for equipment, allowing pass-throughs for capital but controlling all other hospital expenditures. The system has benefited equipment producers, at least in the short run. The expectation was that if costs for labor and services were controlled and capital equipment costs could be passed through, then hospitals would channel funds into capital projects and the purchase of labor-saving equipment. Indeed, a 1988 study found that spending for major movable equipment, in particular, increased after 1983.[15]
Frank A. Sloan, Michael A. Morrisey, and Joseph Valvona, "Effects of the Medicare Prospective Payment System on Hospital Cost Containment: An Early Appraisal," Milbank Quarterly 66 (1988): 191-220.
Debates on how to finally structure capital costs continued through the 1980s. HCFA urged merging capital costs into PPS. ProPAC, however, changed course in 1990 and opposed any change in the capital-cost program. Capital spending continued to rise, jumping 28 percent in 1989 to $15 billion. HCFA approved one-third of that amount, or about $5 billion in Medicare capital spending. This amount represented about 9 percent of the program's $58 billion, part A budget. Estimates for 1990 were $19.3 billion, up another 27 percent.[16]
Stephen K. Cooper, "ProPAC Nixes Capital Expenditure Reform Scheme," Healthweek, 7 May 1990, 15, 36. Any decision on the pass-through was delayed during 1990, and, regardless of the change, it would not take effect until 1992 or 1993. Erich Kirshner, "HCFA, AHA Maneuver on Medicare Capital Reimbursement," Healthweek, 30 July 1990, 9.
PPS could have had a much greater impact on innovative medical technology if the original plan had been fully implemented. Through the 1980s, the capital-cost pass-through operated as a modest safety valve on some innovative devices that are considered capital equipment. Yet, continuation of the debate into the 1990s underscores the pervasive market uncertainty facing medical device producers.
Information through Technology Assessment
Another technique to control the adoption and diffusion of technology is to assess it prospectively. If a new technology does not pass the evaluation screen, a market barrier can be imposed. Ongoing comparative assessment can promote the abandonment of outdated technologies and thereby affect the rate of diffusion.
Prospective assessment is not limited to evaluation on the basis of cost. Indeed, the concept, as originally conceived, was quite broad. The idea was formally developed by Congressman Emilio Daddario, chair of the House Subcommittee on Science, Research, and Development in 1965.[17]
H. David Banta and Clyde J. Beheny, "Policy Formulation and Technology Assessment," Milbank Memorial Fund Quarterly 59 (1981): 445-479.
His work recognized thatscientific and technological developments present potential social consequences. The goal of technology assessment was to examine the safety, efficacy, indications for use, cost, and cost-effectiveness of a particular technology, including the social, economic, and ethical consequences to improve health care decisions.[18]
For a thorough overview of all the forms of technology assessment and the variety of institutions engaged in the enterprise, see the Institute of Medicine, Assessing Medical Technologies (Washington, D.C.: National Academy Press, 1985).
While the concept of medical technology assessment sounds high-minded, its implementation raised a number of very complex issues. Assessment involves three levels of information: gathering of data, evaluating data, and imposing decisions through regulation based upon the evaluation. In addition, assessments can apply to a broad range of technologies—drugs, devices, procedures, and systems—or to just one. Finally, assessment can cover many attributes of a technology, including safety or cost, or it can be limited to only one attribute. Thus, for example, the Food and Drug Administration can be considered a technology assessment agency. Its jurisdiction extends to drugs and devices (not procedures); it evaluates the attributes of safety and efficacy only (not cost or cost-effectiveness); it can require data gathering from the producer; and it regulates based on its evaluation of the data.
It is clear that those who control technology assessments become pivotal gatekeepers for adoption and diffusion of that technology. Thus the history of medical technology assessment is inextricably linked to the tensions among interest groups to influence assessment results. Much of the battle has also involved private sector efforts to prevent government from increasing its share of the gatekeeper function. This concern is especially acute when the goals of the government are to contain costs and arguably to prevent innovations from entering the marketplace, particularly if they are perceived to increase costs.
Government agencies entered the technology assessment process at an early stage to accomplish a variety of goals. As might be expected, government efforts were piecemeal and underfunded. The market does not generate necessary and complete information on medical technologies because clinical testing is time-consuming and expensive. It is easy for competitors to become free riders by observing the technological choices of others. Government plunged ahead. In addition to the FDA, new
programs were created in both the administrative and the legislative branches of government, including the Health Program at the Office of Technology Assessment (legislative branch), the National Center for Health Services Research (NCHSR), the NIH's Office of Medical Applications of Research (OMAR), and, for a short time, the National Center for Health Care Technology (NCHCT). HCFA uses the services of the Offices of Health Technology Assessment (OHTA) within HHS to provide assessment data. OHTA has a very small budget and is limited to safety and efficacy review.[19]
For a discussion of the institutional issues in technology assessment, see Foote, "Assessing Medical Technology."
Both producer and physician groups in the private sector resented government efforts to control technology. Their opposition has been particularly intense when a government agency has the power to regulate decision making rather than the ability just to gather information. For example, in 1978 the American Medical Association (AMA) opposed the creation of the NCHCT, which had regulatory power, alleging that it would interfere with medical practice. HIMA argued that the new assessment agency was a threat to innovation. Both groups strongly and successfully advocated the dismantling of the agency in 1982.[20]
Ibid., 69.
When PPS passed, cost control became an important federal goal. Despite their concerns, technology producers understood that information about benefits as well as costs was now essential for government. A cost-conscious government payer (HCFA) accounted for the direct and the indirect purchasers of over 40 percent of all medical technology. HCFA would gather information regardless of whether the industry or professions cooperated. Government clearly assumed an important and indisputable role. Indeed, as the 1980s progressed, cost dominated the technology assessment debate, and HCFA established itself in a leadership position. Many private payers followed its lead.
Many believed, although HCFA vehemently denied it, that the agency made coverage decisions based predominantly on cost issues, at least since the inception of PPS. The debate about HCFA's jurisdiction and authority to evaluate a technology's cost-effectiveness in order to make coverage determinations illustrates the nature of the debate.
The legislative mandate of HCFA is to decide whether a medical
service is "reasonable and necessary" in order to provide Medicare coverage. Many coverage decisions are made by insurers that contract with Medicare, and there is much regional variation. However, some major coverage decisions are made by the national agency.[21]
See discussion in chapter 4.
In a rule proposed in 1989, HCFA attempted to clarify the meaning of "reasonable and necessary" under the Medicare program. It proposed to add the criterion of cost-effectiveness to considerations of safety and effectiveness; it also proposed to consider whether a technology was experimental or investigational. It justified its proposal thus: "HCFA is including cost-effectiveness as a criterion because we believe considerations of cost are relevant in deciding whether to expand or continue coverage of technologies, particularly in the context of the current explosion of high-cost technologies."[22]54 Federal Register 4302-4318, 4308-4309 (30 January 1989).
Both the AMA and HIMA opposed the addition of cost-effectiveness as a coverage criterion, arguing that it raised substantial legal, methodological, and policy questions. The AMA also argued that HCFA lacked the statutory authority to make the decision and that Medicare's purpose is to meet medical needs, not to make evaluations and comparisons among technologies that amount to the practice of medicine. It also argued that cost-effectiveness analysis is impractical, time-consuming, and inherently subjective.[23]
William McGivney, "AMA Responds to HCFA's Proposed Coverage Criteria and Procedures," American Medical Association Tech 2 (May 1989): 4-6.
HIMA had similar concerns, and added that cost should be a factor only in reimbursement decisions, not for coverage policy. HIMA stated that the proposal would provide a major barrier to entry for new technology. These concerns of organized medicine and industry echo their complaints against the now-defunct NCHCT. No action had been taken on the proposal by 1990. Regardless of the outcome, however, it is likely that HCFA will take cost into consideration, either overtly with clarified authority or somewhat more indirectly.Cost-effectiveness considerations have also crept into other technology assessments. A recent example involves the deliberations of the FDA Advisory Panel on Obstetrics and Gynecology Devices. The FDA has no statutory authority to consider costs in its deliberations; it is limited by law to considerations of safety and efficacy. However, the advisory panel voted unanimously against approval of Healthdyne's home uterine monitoring systems
for premature labor.[24]
Cited in James G. Dickinson, ed., Dickinson's FDA, March 15, 1989, 9.
The device reads the uterine activity of a pregnant woman and transmits it by modem to a professional in a clinical setting. The advisory panel was concerned about the absence of direct clinical proof that product use reduced morbidity or mortality, although others argued that in vitro diagnostics have never been required to produce such data. Also apparent in the advisory panel deliberations were concerns that the technology would become a costly new standard of care. The American College of Obstetricians and Gynecologists (ACOG) issued a statement that the device would cost about $80 a day, averaging $5,616 per patient and potentially costing $5.6 billion a year. Observers commented that costs appeared to have influenced the panel's decision to disapprove the device: "FDA advisory panels are intended to guide the agency on product safety and efficacy and risk-benefit. Increasingly, especially in the area of devices, panels have come under pressure from … [HCFA] … to factor in cost-benefit considerations as well."[25]Ibid.
The trend for the 1990s is movement from traditional technology assessment to the measurement of outcomes. Broadly conceived, outcomes research measures the patient's quality of life as the result of medical treatment. Purchasers are seeking value for their money, and providers are interested in how to ensure the best quality care. While the thrust of outcomes research is slightly different from technology assessment, the barriers to its attainment are similar. There is no consensus on how to measure outcomes or who should pay for it.
Despite these problems, Congress demonstrated its support for the concept of outcomes research with the 1989 creation of the Agency for Health Care Policy and Research (AHCPR) to "enhance the quality, appropriateness, and effectiveness of health care services and to improve access to services."[26]
Public Law 101-239. See also Ron Geigle and Stanley B. Jones, "Outcomes Measurement: A Report from the Front," Inquiry 7 (1990): 7-13. For details on the new agency, see U.S. Department of Health and Human Services, AHCPR: Purpose and Programs (Washington, D.C.: September 1990).
Congress appropriated $568 million over the next five years to fund its activities. Among its goals are the facilitation of practice guidelines to assist physicians, a treatment effectiveness program to assess the effects of variations of treatment on outcomes, and support for programs in health services research and training of researchers.Many express optimism about the potential for outcomes research. But even in its earliest stages, there is mutual distrust
between payers and providers and a belief that payers will assess technology on the basis of cost, not quality. As one observer put it, "[W]hat looks like effectiveness research from one perspective looks like cost-cutting from another. Providers continue to distrust the long reach of government; government looks with narrowed eyes on the amount spent for providers' services."[27]
Janet Ochs Wiener, ed., Medicine and Health: Perspectives, 9 October 1989, 1.
Case Studies: The Impact of Government Cost Containment on Distribution
The introduction of cost containment into government payment policies has had and will continue to have a powerful impact on medical device producers. HCFA is now as important a barrier as the FDA. Medicare coverage and payment policies can delay and limit access of an innovation to the marketplace. Most importantly, market access becomes problematical. There are no clear policy guidelines for producers as HCFA continually experiments with regulatory approaches, and Congress periodically intervenes as a watchdog and accomplice in the search for ways to control government spending. Additionally, there are short-run market distortions based on the idiosyncrasies of the Medicare system. For example, imposing DRGs on the hospitals only opened opportunities in the less regulated outpatient segment of Medicare. How long that differential will last is not known, as extensions of PPS to physicians' fees and outpatient settings were on the horizon in 1990. Once again, the future is uncertain, hampering long-term strategic planning in the industry.
The following case studies illustrate the powerful influence of government payment policies on a new technology. They also introduce the realities of policy proliferation. Cost containment appeared in an already complex, regulated market. In the cases of the intraocular lens and the cochlear implant, the interrelationships of the multiple policies play a role in the potential success or failure of the technologies.
Intraocular Lenses
Millions of Americans suffer from eye diseases that impair vision. Cataracts, opacities of the lens of the eye, often result from
degenerative changes in old age or from diseases such as diabetes. The symptoms include gradual loss of vision, and treatment most commonly involves removal of the diseased lens and the implantation of an intraocular lens (IOL) to restore sight.[28]
Eileen McCarthy, Robert Pokras, and Mary Moien, "National Trends in Lens Extraction: 1965-1984," Journal of the American Optometric Association 59 (January 1988): 31-35.
Ophthalmology in general, and IOLs in particular, represent one of the largest and most dynamic health care markets. The FDA regulates IOLs, and, because most of the implant candidates are elderly, the market is strongly tied to Medicare payment policy. This policy, as well as the interaction between regulation and reimbursement, has the potential for significant impact on the industry.
IOLs are one of the few ophthalmic products that the FDA has placed in Class III.[29]
See chapter 4 for a detailed discussion of FDA regulation.
Regulated since 1979, IOLs are subject to a special requirement imposed by Congress and enforced by the FDA.[30]David M. Worthen et al., "Update Report on Intraocular Lenses," American Academy of Ophthalmology 88 (May 1981): 381-385; and Walter J. Stark et al., "The Role of the Food and Drug Administration in Ophthalmology: An Editorial," Archives Ophthalmology 104 (August 1986): 1145-1147.
As with all Class III devices, the FDA reviews data on safety and efficacy in the premarket approval (PMA) application. During the experimental stage, Class III products may receive an IDE, or investigational device exemption, that allows them to be used in controlled studies while the manufacturer gathers and evaluates the data about their safety and efficacy. The collection of data supporting a PMA is expensive and time-consuming and may represent a significant barrier to entry for smaller innovative firms. For IOLs, however, a special exception was made whereby the producers could charge for the costs of the implanted lenses while still in the investigational (IDE) stage. This exemption facilitated the development of IOLs during the two or more years of FDA-required device testing in clinical settings.Indeed, the availability of Medicare payment for this primarily elderly patient population essentially guaranteed a large, stable market for lens removal and IOL implantation. The average cataract patient is sixty-eight years old; Medicare is the sole payer for almost all cataract surgery. The frequency of IOL implants has grown rapidly in the 1980s. In 1979, there were 177,000 implants; by 1986, the number was 888,000. By 1988, there were 1.2 million implants in the United States and another one million internationally. Annual IOL sales have been estimated at $360 million in the United States alone (see table 9).[31]
Biomedical Business International 12 (17 May 1989): 70-71.
Medicare pays close to $1.5 billion annually for cataract operations,
| ||||||||||||||||||||||||||||||||||||
making them the largest item in the Medicare program in 1988.[32]
Milt Freudenheim, "Medicare's Curbs on Cataract Fees," New York Times, 15 November 1988, C2.
During the 1980s much of the treatment shifted from hospital to outpatient surgery centers or physicians' offices, which are covered under Part B of Medicare and thus are not under the DRG system. The growth can be attributed to advances in the technology, including cataract management, anesthesia, surgical technique, and postoperative care. New IOL technology includes soft lenses that can be implanted with smaller incisions (the one-stitch lens is a recent innovation), and bifocal implants and other specialty lenses have been developed recently.The industry is dynamic and competitive. There are a number of companies in the IOL field, ranging from very large firms such as Johnson & Johnson (IOLAB), CooperVision (recently acquired by Alcon/Nestle), and Allergan (purchased by SmithKline Beecham in 1989). Smaller firms include IOPTEX Research, a privately held industry leader, and Chiron Ophthalmics, a subsidiary of Chiron Corporation, a biotechnology firm. There are also foreign IOL makers from West Germany, France, Belgium, Israel, and Japan.
Changes in both Medicare policy and FDA regulations present threats to the IOL market. Congress has reduced federal
Medicare payments for cataract surgery twice since 1986. HCFA has lowered the amount of payment to physicians for the procedure. These changes have come in the wake of allegations of market abuse by cataract surgeons. Congress investigated the situation as early as 1985;[33]
House Ways and Means Committee, Medicare Reimbursement for Cataract Surgery 99-37, 99th Cong. (Washington, D.C.: GPO, 1 August 1985).
additional hearings were held in 1990, prompted by reports that surgeons employed abusive marketing techniques to round up elderly patients, and that many earned more than $1 million a year performing surgery covered by Medicare.[34]Dwight E. M. Angell, "Cataract Surgeons Face a Critical Eye," Healthweek, 23 April 1990, 1, 8-9.
In 1990, HCFA reduced the payment rate for an IOL implanted during cataract extraction. The revised rate of $200 meant an average reduction of at least $100 from the former IOL rate.[35]
Freudenheim, "Medicare's Curbs."
This lower rate was based on an audit by the Office of the Inspector General to determine how much was actually paid for lenses after subtracting various rebates and discounts often associated with lens purchases. However, many of the newer specialty lenses average over $300 each.The FDA has imposed new requirements for data collection as well. For new bifocal and multifocal products, fifty implants have to be studied for a year, and then the studies can be expanded to five hundred implants. Overall, it is likely to take nearly four years for a new lens to receive premarket approval. The longer period for approval is less onerous if the innovator receives at least partial payment for the experimental lens implants. Rumors have been circulating that the exemption allowing payment for IOLs under IDEs will soon be rescinded. HCFA officials state that this move will encourage producers to progress from the IDE to the PMA stage. They assert that companies have been allowing products to languish under IDEs because the economic incentive to go to market is reduced by the exception. The device is paid for in either case. However, the longer testing requirements and the threatened withdrawal of payment during the investigational period potentially will have a significant impact on newer, less well-capitalized entrants.
Cochlear Implants
Cochlear implants, a technology that permits individuals with profound hearing loss to receive auditory cues, have had a very
different reception than IOLs. Unlike implanted lenses that diffused rapidly to millions of elderly, cochlear implants have not fared well. Indeed, Medicare reimbursement was made for only sixty-nine such implants in fiscal 1987, despite estimates that sixty thousand to two hundred thousand Americans could benefit from the device. Many industrial competitors never entered the field or have since abandoned it, leaving only three firms still in the market in 1990. This medical device has met resistance throughout its history, and the collective impact of policy hurdles has been profound.
The cochlea, a structure in the inner ear, translates sounds from mechanical vibrations to electrical signals. These signals are produced by cells in the cochlea. The cells have a fringe of tiny hairs that bend in response to vibrations in the outer and middle ear. Those responses produce electrical signals that stimulate the auditory nerve and send messages that the brain interprets as sound.[36]
Nancy M. Kane and Paul D. Manoukian, "The Effect of the Medicare Prospective Payment System on the Adoption of New Technology: The Case of Cochlear Implants," New England Journal of Medicine 321 (16 November 1989): 1378-1383.
This natural system of hearing is versatile enough to transmit the full range of sounds. If the hair cells in the cochlea are damaged by injury or disease, the individual is condemned to deafness. Over two hundred thousand Americans suffer this profound hearing loss, and conventional hearing aids are useless for them. Cochlear implants, at least at this stage of development, cannot restore the world of sound. They do allow for reception of sounds such as sirens and voices, auditory cues that are vital for safety and for some social interactions. But the recipient of the implant cannot hear normal conversation.
The possibility of producing useful hearing by electrical stimulation of the cochlea occurred by accident.[37]
Robin P. Michelson, "Cochlear Implants: Personal Perspectives," in Robert A. Schindler and Michael M. Merzenich, eds., Cochlear Implants (New York: Raven Press, 1985), 9-11.
When an amplifier used in the operating room to monitor the cochlear response oscillated, the patient heard a very high-pitched tone. Some early work on this type of induced stimulation was published in 1955 and 1956.[38]William F. House, "A Personal Perspective on Cochlear Implants," in Schindler and Merzenich, Cochlear Implants, 13.
Researchers undertook additional work during the early 1960s, but they encountered significant problems. Among the barriers were included adverse patient reactions to the insulating silicone rubber used in the first primitive devices and concern about the effects of long-term stimulation on all auditory sensation. In 1965, the results of studies were submittedto the American Otological Society but were rejected as too controversial for presentation.
As implant technology improved in the late 1960s, some of the problems were resolved, but concerns over ethics and long-term effects still dogged the technology. The National Institutes of Health did not provide any funding for the scientific research, a refusal that some scientists in the field attributed to the bias against biomedical engineering among NIH peer review groups.[39]
See the discussion in chapter 3 on an antiengineering bias at the NIH.
Several policy breakthroughs occurred in the 1970s, when the NIH focus began to shift toward goal oriented, or targeted, programs that would produce identifiable results. The NIH established an intramural program to investigate cortical and subcortical stimulation, primarily for blindness but also for other neurological disorders. While hearing stimulation was not an important part of the original program, it became of greater interest when the research on cortical visual implants appeared clearly unsuccessful. In 1977, the NIH instituted an independent assessment of patients with cochlear implants. The Bilger Report, produced at the University of Pittsburgh, concluded that these products were a definite aid to communication.[40]
R. C. Bilger et al., "Evaluation of Subjects Presently Fitted with Implanted Auditory Prostheses," Annals of Otolaryngology, Rhinology and Laryngology, supplement 38 (1977) 86: 3-10. Discussed in F. Blair Simmons, "History of Cochlear Implants in the United States: A Personal Perspective," in Schindler and Merzenich, Cochlear Implants, 1-7.
Seven years later, in November 1984, the FDA approved the design of a single-channel device for cochlear implantation. The 3M Company produced the device in conjunction with Dr. William House, an early researcher and chief inventor. The device consisted of a receiver similar to that of a hearing aid, a speech-processing minicomputer that transforms the sound signals into electrical signals, another receiver for those electrical signals that is implanted under the skin above and behind the ear, and a thin wire inserted surgically through the mastoid bone into the cochlea to transmit the signals (see figure 23).
By 1985 a handful of companies had entered the market. The 3M Company was the clear leader with the only approved product. Others included the Nucleus Group, an Australian company and parent of the U.S. Cochlear Corporation, Biostim, and Symbion, an outgrowth of research at the University of Utah.[41]
Biomedical Business International 8 (29 March 1985): 47-48.
A private industry group reported that leading hearing-aid manufacturers did not enter the marketplace because "the FDA-related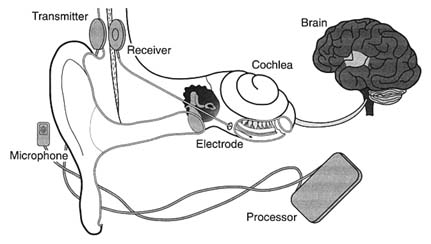
Figure 23. The cochlear implant.
Source: The 3M Company, Cochlear Implant System, n.d.
expense of developing and testing such devices is prohibitive." It was reported that 3M had budgeted over $15 million for cochlear implant development.
Once approved by the FDA, the device faced the hurdle of Medicare's coverage and payment decision. After several years of deliberation, and following endorsement of the device by the AMA in 1983 and the American Academy of Otolaryngology in 1985, Medicare issued a favorable coverage ruling for both single-channel and multichannel devices in September 1986. The next step was for HCFA to assign the technology to a code which would then provide the means for establishing appropriate levels of payment for procedures.[42]
Kane and Manoukian, "The Effect of Medicare," 1379.
HCFA has considerable discretion in the placement of a new procedure into the DRG system. If the device is assigned to a DRG that does not cover the cost during the diffusion period, hospitals implanting the devices will lose money. Hospitals that increase the proportion of cases involving these devices lower their operating margins in those DRGs. ProPAC recommended in 1987 that the cochlear implant be assigned to a device specific, temporary DRG. HCFA did not follow ProPAC's recommendation. Instead, in May 1988 the agency announced a DRG placement
that would not pay the full estimated cost of $14,000 for the implantation of the device.[43]
Ibid., 1380.
Evidence accumulated that hospitals had a strong disincentive to provide cochlear implantation. Ten percent of the 170 hospitals involved openly acknowledged to researchers that they restricted implantations because of the loss of $3,000 to $5,000 for each Medicare case.[44]
Ibid., citing Cochlear Corporation personal communication.
These policies limited the size of the market and deeply affected the private sector producers. The 3M Company stopped marketing the single-channel model actively and halted research on multichannel devices because of the low use rate of both models. The small market discouraged additional investment. There were five firms that developed cochlear implants for the U.S. market from 1978 through 1985. By 1990, three had left and there were no new entrants with FDA approved devices.[45]
Ibid.
It was undisputed that this new technology had limitations, but it also was recognized as useful and beneficial to certain classes of patients. The policy environment was relatively unresponsive to early development. The quest for FDA approval was difficult, time-consuming, and costly. The response of HCFA to the technology was definitely obstructionist. The future of the research and development of this area of technology is now in doubt.
The primary effect of the payment policy has been uncertainty in the marketplace. Firms can no longer count upon growth in their particular market segment. Incremental policy changes are frequent and can have catastrophic effects on the markets for some products. In addition, cost-containment policies have introduced new hurdles to market access, which cause higher costs and delays even for successful new entrants.
Cost control has become an important value in the distribution of medical devices. It presents significant problems because the costs of a new technology are difficult to predict before distribution. Some products may have cost-reducing potential that is not known in the early stages or additional beneficial applications that will emerge during use. It is legitimate to ask how much cost should matter and who should decide that issue.
In addition, the case studies illustrate how complex the policy environment was in 1990. There are significant hurdles at virtually
every stage of innovation. Even promoters such as NIH can place barriers in the paths of the innovators. NIH disapproval can act as a deterrent, as the early years of cochlear implant development reveal. HCFA, once a source of nearly unlimited funds, can significantly delay or even bar technology from the marketplace.
Our medical device patient is now a confirmed recipient of polypharmacy, as the prescriptions have proliferated over time. Before we turn to the prognosis, however, it is necessary to look at the international marketplace. Does the world market provide an outlet for manufacturers constrained by cost controls in the United States? Or are international firms a competitive threat both in the United States and abroad?