5
Government Inhibits Medical Device Discovery: Regulation
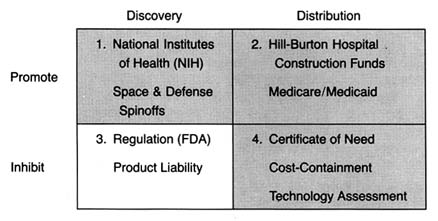
Figure 16. The policy matrix.
The policies discussed in chapters 3 and 4 changed American medicine. Neither was designed specifically to promote the medical device industry; federal R&D was primarily directed at bio-medical research, and federal and state payment programs were designed to increase access to health care. Nevertheless, the medical device industry benefited. Increasingly sophisticated devices became available to large numbers of people. Devices began to lose their association with quack products and were frequently linked to therapeutic breakthroughs.
However, many of these sophisticated new technologies, such as pacemakers, kidney dialysis equipment, and diagnostic instruments, presented risks along with their potential benefits. Adverse effects associated with cardiac pacemakers, intrauterine
devices (IUDs), and implanted interocular lenses, in particular, raised public concern.
The ground was fertile for federal regulation of these products. The late 1960s and the 1970s were a time of growing consumer activism and power. Product safety became a desirable value that the federal government was expected to protect. The government was expanding safety regulation in many new areas, including consumer products, the workplace, and the environment. In many instances, Congress created new regulatory agencies to address safety issues, including the Environmental Protection Agency (1969), the Occupational Safety and Health Administration (1972), and the Consumer Product Safety Commission (1972). In the case of medical devices, the FDA was already in place with expertise and some preexisting jurisdiction over the industry.
This chapter describes the political process leading to the expanded regulation of medical devices, the first major legislation directed exclusively at the device industry. (See figure 16.) Issues related to the structure and implementation of the Medical Device Amendments of 1976 are discussed, illustrated by cases such as IUDs, lithotripsy equipment, pacemaker components, and tampons.
Once again, a brief caveat before proceeding. By the 1970s, the effects of policy proliferation became apparent. For example, the impact of some regulation was blunted by the Medicare policies that encouraged purchases regardless of cost. Thus costs associated with regulation could be passed along without concern. For other products, the interaction with regulation and product liability increased environmental threats. Issues relating to product liability are discussed fully in chapter 6.
Medical Device Regulation Comes of Age
The Limitations of FDA Authority
As we saw in chapter 2, the FDA had very limited powers over medical devices under the 1938 Food, Drug, and Cosmetic Act. Its primary authority was seizure of individual devices found to be adulterated or misbranded. Most of the FDA's early enforcement
activity was directed toward controlling obvious quack devices. However, even this limited regulatory activity declined during World War II. Because of the scarcity of metals and other critical materials, production of nonessential devices was restricted, and consequently the pace of seizures and prosecutions dropped to less than six per year.[1]
House Committee on Government Operations, Hearings on Regulation of Medical Devices (Intrauterine Contraceptive Devices), 93rd Cong., 1st sess. (Washington, D.C.: GPO, 1973), 180.
After World War II, there was an increase in device quackery because of the cheap availability of war surplus electrical and electronic equipment.[2]
Davidson, "Preventive 'Medicine' for Medical Devices: Is Further Regulation Required?" Marquette Law Review 55 (Fall 1972): 423-424.
A variety of products that used dangerous gases (such as ozone and chlorine), radio waves, heat, and massage were marketed for the treatment of almost every disease known. Among the most dangerous were quack devices that used radium, uranium ore, and other radioactive substances and that purported to cure common problems such as sinus infections and arthritis.The pace of FDA seizures picked up in response. In a report in 1963, the Bureau of Enforcement of the FDA's Device Division stated that from 1961 to 1963, the FDA seized 111 different types of misbranded or worthless devices, involving 15,070 individual units. Fifty-four diagnostic and treatment devices were taken in 358 seizure actions from June 1962 to June 1963.[3]
Milstead, 1963 Congress on Quackery, 30.
Some states tried to tackle the problem with their own legislation. For example, California had passed a state Pure Foods and Drug Act in 1907, one year after the first federal act. The law prohibited any claim for a food, drug, or device that was false or misleading in any particular. California brought over sixty court actions from 1948 to 1957. The focus of this early activity was on fraudulent devices; concern about the safety of clearly therapeutic products came later.[4]Ibid.
Congressional legislative activity focused on drug risks in the late 1960s. Congress increased FDA power to regulate drugs in 1962 in response to controversial evidence linking birth defects and the drug thalidomide.[5]
Temin, Taking Your Medicine, 123-126.
The 1962 amendments to the Food, Drug, and Cosmetic Act greatly strengthened federal power over drugs by requiring proof that the product was both safe and efficacious before it received FDA marketing approval. Because of the previous distinction between drugs and devices made in the 1938 law, these expanded powers did not apply to new medical devices.[6]See discussion in chapter 2.
By the late 1960s, problems associated with legitimate, therapeutically desirable medical devices that were flooding the marketplace began to surface. In 1970, Dr. Theodore Cooper, then director of the NIH Heart and Lung Institute, completed a survey of the previous ten years that revealed 10,000 injuries from medical devices, including 731 deaths. Defective heart valves caused 512 of the deaths.[7]
U.S. Department of Health, Education, and Welfare, Cooper Committee, Medical Devices: A Legislative Plan, Study Group on Medical Devices (Washington, D.C.: GPO, 1970). Cited and discussed in Medical Device Amendments of 1975, Hearings on H.R. 5545, H.R. 974, and S. 510 Before the Subcommittee on Health and the Environment of the Committee on Interstate and Foreign Commerce, statement of Rep. Fred B. Rooney, 94th Cong., 1st sess., 199. See also Theodore Cooper, "Device Legislation," Food, Drug, Cosmetic Law Journal 26 (April 1971): 165-172. There have been challenges to the data in the Cooper report, but the public attention the study received made the issue of device safety politically salient.
In particular, problems had arisen involving defective pacemakers[8]U.S. Comptroller General, Food and Drug Administration's Investigation of Defective Cardiac Pacemakers Recalled by the General Electric Company 21 (1975). GE decided to voluntarily recall over 22,000 pacemakers because some malfunctioned due to moisture that seeped into the pacemaker circuitry, probably due to faulty seals.
and intrauterine devices.[9]See discussion in this section.
Congress, the FDA, and the public became concerned.Creative Regulation by the FDA
The increase in legitimate medical devices complicated the FDA's regulatory efforts. The sophisticated new technologies, such as pacemakers, kidney dialysis units, cardiac, renal, and other catheters, surgical implants, and diagnostic instruments, challenged the FDA's expertise. The agency's regulatory power was limited to seizure of products already on the market. In order to bring a seizure action under the law, the FDA had to consult experts, sponsor research, and gather data to meet its statutory burden of proof in court. The real problem was that seizures were simply not a reasonable response to devices that had benefits as well as risks. The goal was not to remove these products from the market as much as to ensure that these innovations were safe.
In the absence of additional authority, the FDA began to implement the law more aggressively. One tactic was to construe the statutory definition of drug broadly enough to include products that were clearly medical devices and thus would allow the agency to regulate devices much as it regulated drugs.
Device companies challenged this effort to impose drug regulation on devices. Two important court decisions in the late 1960s upheld the FDA's broad reading of the term drug . In AMP v. Gardner , the court reviewed the FDA's classification as a "new drug" a nylon binding device used to tie off severed blood vessels during surgery.[10]
389 F.2d 825 (2d Cir. 1968).
The court broadly construed the purpose of the 1938 act and the 1962 amendments, holding that the goal was to keep inadequately tested and potentially harmful "medical products" out of interstate commerce. Emphasizing the protectivepurposes of the law enabled the government to regulate as a drug any product not generally recognized as safe.
The next year, the Supreme Court followed similar reasoning in United States v. An Article of Drug … Bacto-Unidisk .[11]
394 U.S. 784 (1969).
In 1960, after the product had been in use for four years, the secretary of HEW classified an antibiotic disk as a drug. The product, which never came into contact with the human body and was therefore not metabolized, was used as a screening test in a laboratory to determine the proper antibiotic to administer. Its classification as a drug came after the agency received numerous complaints from the medical profession, hospitals, and laboratory technicians that the statements of potency for the disks were unreliable. The FDA found it "vital for the protection of public health" to adopt the regulations.[12]25 Federal Register 9370 (30 September 1960).
The Court, acknowledging that the FDA was an expert agency charged with the enforcement of remedial regulation, deferred to the secretary's medical judgment. It concluded that the term drug was a legal term of art for purposes of the law, which was a broader interpretation than the strict medical definition. The Court determined that the parallel definitions of drugs and devices, discussed previously, was "semantic." Concluding that there was no "practical significance to the distinction" until subsequent amendments to the 1938 Act, the Court gave the term "a liberal construction consistent with the act's overriding purpose to protect public health."[13]
394 U.S. 784, 798 (1969).
IUDs
The FDA's response to problems associated with implanted intrauterine devices illustrates its efforts to regulate creatively to protect the public. IUDs to prevent pregnancy had been available since the turn of the century. Although they had been generally dismissed as dangerous by respectable practitioners, the technology began to be reevaluated in the late 1950s. The renewed interest in contraceptives, the availability of inert plastics that caused fewer tissue reactions, and the growing controversies about the safety of the new contraceptive pills all encouraged research on IUDs.
IUD devices began to enter the market in the mid-1960s.
From 1969 to the early 1970s, IUD use skyrocketed. By the end of 1970, three million women in the United States had been fitted with a variety of IUD devices. Marketing was aggressive, and the competition among firms was keen. The top sellers included Ortho Pharmaceutical's Lippes Loop, the Saf-T-Coil produced by Schmidt Labs, and the now infamous Dalkon Shield introduced by A. H. Robins in January 1971.[14]
The Dalkon Shield claimed to be superior to other products because of its unique shape. The nature of the product and the harms related to its design are discussed more fully in chapter 6.
G. D. Searle entered the market in the same year with the Cu-7, a device shaped like the number 7 with a small thread of copper wound around the vertical arm, which the company claimed increased the product's efficacy. The data regarding unit sales testify to the early success of these products (see table 6).Despite burgeoning sales, product safety remained a concern. As early as 1968, an FDA advisory committee on obstetrics and gynecology cited significant injuries and some deaths associated with IUDs.[15]
House Hearings on Medical Devices, Advisory Committee on Obstetrics and Gynecology of the Food and Drug Administration, Report on Intrauterine Contraceptive Devices (1968), 441.
The devices generated numerous complaints, and there were reports of infections, sterility, and, on some occasions, death.Under the device provisions of the 1938 law, the FDA had only the limited power to seize individual products, an impractical remedy in this situation. An internal FDA memorandum recommended that the AMP v. Gardner precedent be used to designate all products intended for prolonged internal use to be considered "drugs" for purposes of premarket approval.[16]
House Hearings on Regulation of Medical Devices, memorandum of William Goodrich, assistant general counsel of the FDA, 19 March 1968, 205-206.
If this recommendation had been adopted, all implanted devices, including pacemakers and IUDs, would have been officially considered drugs under the law.The agency did not go so far. In 1971 it considered a proposed rule on the classification of IUDs. At that time, G. D. Searle began to market the Cu-7 IUD. Because this device contained a noninert substance, the FDA's final rule in 1973 distinguished between device-type IUDs and drug IUDs. The agency treated as a regulated drug any IUD that contained heavy metals or any substance that might be biologically active in the body. An IUD was a device, hence exempt from premarket approval requirements, if it was fabricated entirely from inactive materials or if substances "added to improve the physical characteristics [did] not contribute to contraception through chemical action on or within the body."[17]
44 Federal Register 6173 (31 January 1979).
Thus, Searle's Cu-7 was subjected
| ||||||||||||||||||||||||
to premarket approval, as was Progestasert, an IUD with a timed-release contraceptive hormone that entered the market in 1976.
FDA officials later admitted the frustration of using the drug provisions as a substitute for adequate device regulation. Despite efforts to increase the staff for the Medical Devices Program, the agency had limited resources for the regulation of devices as drugs.[18]
House Hearings on Regulation of Medical Devices, 183.
Other important jurisdictional issues arose as well. In 1972, Peter Barton Hutt, general counsel for the FDA, said that "The administrative burden of handling all devices under the new drug provisions of the act would be overwhelming…. If we were to reclassify all devices as new drugs, difficult legal issues would be raised about our authority to allow them to remain on the market pending approval of an NDA [new drug approval]. Wholesale removal of marketed products would, of course, not be medically warranted."[19]House Hearings on Regulation of Medical Devices, statement of Peter Barton Hutt, 209.
Without clear legislative authority, the FDA was unwilling to regulate devices through use of the provisions intended to apply only to drugs.As might be expected, problems related to devices continued to arise. When reports of severe adverse reactions were specifically associated with the Dalkon Shield, an advisor to the FDA recommended that it be removed from the market. The big question was how to do so under the law. The only power the FDA had was to get a court injunction to halt interstate shipments of adulterated and misbranded products and to proceed to seize them one at a time. Of course, for implanted products, safety would clearly be better served by preventing these products from entering the market in the first place. A. H. Robins finally admitted the inevitability of some government action,
and, in the wake of significant adverse publicity, the company voluntarily suspended sales, pending a hearing of FDA advisory bodies. The product never returned to the market.[20]
Enter policy proliferation. The company's action may well have been motivated primarily by the fear of lawsuits, not the fear of FDA action. In any event, the FDA had few powers to invoke. Issues relating to product liability will be discussed in chapter 6.
The FDA ultimately got the outcome it desired, but its formal regulatory impotence did not go unnoticed by Congress or the public.Congress Takes Action
The limited powers of the FDA had been graphically demonstrated during the Dalkon Shield controversy. Congress not only was aware of the publicity concerning harmful devices but also had held hearings on several products during this period.[21]
Pacemaker hearings, medical device (IUD) hearings.
Bills to expand the FDA's authority over devices had been introduced every year from 1969 to 1975. The likelihood of congressional action was increased by the fact that consumer activism was at its peak. The controversies surrounding IUDs mobilized the nascent women's movement, and defective cardiac pacemakers caused concern among the elderly. Ralph Nader's Health Research Group vigorously lobbied government to protect consumers in the areas of medicine and health products.On the other side, the medical device industry was not well organized. Until this time, government had been either neutral or a benefactor, not a threat to the industry's well-being. In fact, there was no trade association until the mid-1970s. Unlike the drug industry, which was represented by the old and powerful Pharmaceutical Manufacturers Association (PMA), the device producers were a disparate group with no clearly identifiable or shared issues. Many were small innovators with little or no experience with the political process.
The prospect of regulation spurred organizing efforts. The Health Industry Manufacturers Association (HIMA) formed in 1976, but it was too late to stop Congress from regulating the industry. Indeed, the organization was established in direct response to the new regulatory threat. Some larger device companies had their own Washington offices that handled government relations; for smaller companies, HIMA was the only representation. HIMA has since become larger and more active, but in the 1970s members of the industry were reactive, not proactive. Regulation was only a matter of time.
The Medical Device Amendments of 1976
The Medical Device Amendments of 1976 sought to provide "reasonable assurance of safety and effectiveness' for all devices.[22]
Public Law 94-295, 90 Stat. 539 (1976) codified at 21 United States Code secs. 360c-360k (1982), (a)(1-3). For detailed discussion of the Medical Device Amendments, see Foote, "Loops and Loopholes"; David A. Kessler, Stuart M. Pape, and David N. Sundwall, "The Federal Regulation of Medical Devices," New England Journal of Medicine 317 (6 August 1987): 357-366; and Jonathan S. Kahan, "The Evolution of FDA Regulation of New Medical Device Technology and Product Applications," Food, Drug, Cosmetic Law Journal 41 (1986): 207-214.
The FDA was to determine whether such assurance existed by "weighing any probable benefit to health from the use of the device against any probable risk of injury or illness from such use." The law conferred powers upon the FDA to regulate medical devices during all phases of development, testing, production, distribution, and use.In order to accomplish these goals, Congress devised a complicated regulatory scheme. This complexity arose from both the diversity of the products to be regulated and the lack of trust between Congress and the FDA at that time. The diversity of devices dictated a regulatory system that would provide levels of government scrutiny appropriate to the nature of each device. The lack of trust meant that Congress did not give the agency discretion to implement the law; instead, detailed provisions were intended to force the agency to regulate with vigor.[23]
See letter from Representative Paul Rogers, one of the authors of the legislation to Alexander Schmidt, commissioner of the FDA, 21 June 1976 (cited in Foote, "Administrative Preemption," 1446, n. 74).
In the law, Congress used two different methods to group medical devices: first, devices were divided into three classes on the basis of risk, with increasing rigor from Class I to Class III; and second, they were divided into seven categories (preamendment, postamendment, substantially equivalent, implant, custom, investigational, and transitional). It is not surprising that a complicated system emerged from these numerous divisions.
In brief, Class I, general controls, is the least regulated class, and it requires producers to comply with regulations on registration, premarketing notice, record keeping, labeling, reporting of adverse experiences, and good manufacturing processes. These controls apply to all three classes of devices. Manufacturers of Class I devices must register their establishments and list their devices with the FDA and notify it at least ninety days before they intend to market a device. Tongue depressors are an example of a Class I device. Class II devices are those for which general controls are considered insufficient to ensure safety and effectiveness and for which information exists to establish performance standards. Well over half of the devices on the market are in Class II.
Class III consists of those devices for which general controls alone are insufficient to ensure safety and efficacy and for which information does not exist to establish a performance standard and the device supports life, prevents health impairment, or presents a potentially unreasonable risk of illness or injury. Only those devices placed in Class III receive premarket reviews similar to those conducted on drugs. The manufacturer must submit a premarket approval application (PMA) that provides sufficient data to assure the FDA that the device is safe and efficacious. Only a small fraction (about 8 percent) of all devices are placed in Class III, including heart valves and other implanted products.
The categories set forth in the law established guidelines for classification. For example, implanted devices are assumed to require a Class III placement, and custom and investigational devices can be exempt from premarket testing and performance standards. Examples of implants include cardiac pacemakers and artificial hips; custom devices include dentures and orthopedic shoes; and at present investigational devices include the artificial heart and positron emission tomography (PET) imaging machines. Transitional devices are those regulated as drugs (such as copper-based IUDs) before the passage of the law, and they are automatically assigned to Class III. Devices on the market at the time the law was passed are referred to as preamendment or preenactment devices . These products are assumed to be in Class I unless their safety and efficacy cannot be ensured without more regulation. Manufacturers can petition for reclassification under certain circumstances.
One provision has assumed a greater significance in practice than was perceived when the law was drafted. New devices that are shown to be "substantially equivalent" to a device on the market before the law was passed are assigned to the same class as their earlier counterparts, and manufacturers have to provide information on testing and approval only if the earlier products required it. To receive the designation of substantial equivalence, section 510k requires producers to notify the FDA at least ninety days before marketing. This premarket notification must contain enough information for the FDA to determine whether the device is substantially equivalent to a device already being
marketed.[24]
The process is referred to as a 510k after the number of the provision in the bill.
A product need not be identical, but it cannot differ markedly in design or materials. If a device meets the equivalence requirement, it can go directly to market without further scrutiny. The benefits to manufacturers of "a 510k" are enormous, as their products can enter the market quickly and without great effort.This complex regulatory framework invites maneuvering on the part of producers. Unlike the drug law that treats all new chemical entities (NCEs) alike, the medical device amendments present a large number of options and opportunities to manipulate the system. The FDA's management of the device law has been very controversial. Frequent hearings and investigations by Congress have tended to conclude that the FDA has not measured up.[25]
For example, Report of the House Subcommittee on Oversight and Investigations, Committee on Energy and Commerce, MedicalDevice Regulation: The FDA's Neglected Child (Washington, D.C.: GPO, 1983); Comptroller General Report to Congress, Federal Regulation of Medical Devices—Problems Still to Be Overcome, GAO-HRD-83-53 (Washington, D.C.: General Accounting Office, September 1983); United States General Accounting Office, Report to the Chairman, Senate Committee on Governmental Affairs, Early Warning of Problems Is Hampered by Severe Underreporting, GAO-PEMD-87-1 (Washington, D.C.: General Accounting Office, December 1986); and General Accounting Office, Briefing Report to the Chairman, House Subcommittee on Health and the Environment, Committee on Energy and Commerce, Medical Device Recalls: An Overview and Analysis 1983-1988, GAO-PEMD-89-15r (Washington, D.C.: General Accounting Office, August 1989).
On the other hand, some in industry have accused the FDA of overregulation, inefficiency, and harassment. For its part, the FDA claims that limited resources and expanding demands hamper enforcement.Implementation of the FDA's New Powers
FDA authority over the device industry falls into two general categories—barriers to market entry (premarket controls) and the power to oversee production of a marketed product or to remove it from the marketplace (postmarket controls). Issues of implementation have arisen in both categories.
Premarket Controls: Problems of Classification and Categorization
The classification of the device determines the level of scrutiny it receives. The ability of the law to reduce risks depends upon a rational classification process. If barriers are too high, desirable innovations will be discouraged. If they are too low, the public will not receive the protection the law intended. Given the number of vastly different devices subject to regulation and the limited resources and energy of the agency, there are many problems regarding classifications.
Because the degree of regulation varies significantly depending on the classification of a device, it is not surprising that there
have been disputes over how the FDA evaluates industry petitions to reclassify a device. An important set of judicial opinions clarified the FDAs authority to deny reclassification petitions.
Two appellate court decisions in the D.C. Circuit Court of Appeals affirmed the FDA's discretion to deny reclassification petitions if it finds insufficient scientific evidence to do so. In Contact Lens Manufacturers Association v. FDA,[26]
766 F.2d 592 (1985).
the trade association challenged the FDA's refusal to reclassify rigid gas-permeable lenses (RGP) from Class III to Class I. Hard contact lenses (polymethylmethacrylate, or PMMA) have been marketed in the United States since the early 1950s. Soft lenses (hydroxyethylmethacrylate, or HEMA) lenses are a more recent development. In September 1975, citing their "novelty," the FDA announced that all HEMA lenses would be regarded as "new drugs" and regulated as such. Upon passage of the Medical Device Amendments, all devices regulated as drugs were automatically in Class III under a "transitional" device provision.[27]21 U.S.C. sec. 360j(1)(1)(E).
In 1981, the FDA considered reclassification of RGP lenses, a type of soft lens, into Class I, which would greatly have reduced regulatory oversight. However, after receiving extensive comments, the FDA withdrew its proposed reclassification,[28]48 Federal Register 56, 778 (1983).
and the industry association petitioned the court for review of the FDA's power. The court upheld the FDA's authority to withdraw its proposal. In General Medical v. FDA[29]General Medical v. FDA, 770 F.2d 214 (1985).
the court upheld the FDA's decision to deny a petition for reclassification of the Drionic device, a product used to prevent excessive perspiration.Additional problems have arisen regarding devices on the market before the passage of the law. Many of these devices are considered Class III, but they have been largely ignored. The first premarket approval application (PMA) for a preenactment device was not required until June 1984, a full eight years after the law had been passed. The FDA stated that the implanted cerebellar stimulator was chosen because of contradictory information about its effectiveness for some indications.[30]
The device is used to electrically stimulate the cerebellar cortex of a patient's brain in treatment of intractable epilepsy and some movement disorders.
In 1983, the FDA published a notice of intent to require premarketing approval of twelve other preenactment devices.[31]Medical Device Bulletin (Washington, D.C.: FDA, August 1984). See also Kessler et al., "Federal Regulation," 362, nn. 5, 12.
After years of controversy, the FDA finally required PMAs for preenactment heart valves in June 1987.The FDA has also been criticized for its failure to implement
the Class II requirements. Class II devices are supposed to meet performance standards to ensure safety and efficacy. The statutory provisions for the selection of a standard-setting body and the drafting of standards are exceedingly detailed.[32]
The excessive detail in the law derives from a congressional desire to limit FDA discretion by carefully spelling out procedures to be followed. This strategy is ineffective, as it has hampered the implementation of many provisions.
The process itself would be costly and slow, arguably locking in the state of the art at the time a standard was set. More than 50 percent of the 1,700 classified types of devices are in Class II, but not one performance standard had been issued by 1988.[33]Kessler et al., "Federal Regulation," 362.
Given that performance standards are the only distinction between Class II and Class I, this situation makes a mockery of the classification system.Premarket Notification: Pacemaker Leads
Problems related to pacemaker leads illustrate the controversy surrounding 510k, the provision for premarket notification as a substitute for FDA review. Pacemaker leads connect a pacemaker's power source to the heart muscle itself. Innovators have had many problems with lead design—leads tend to become dislodged and render the pacing device ineffective. Many new designs emerged as the pacemaker industry developed, and there were a significant number of lead failures.
Congress held hearings in 1984 on the Medtronic polyurethane pacemaker leads. The congressional inquiry was prompted by reports that certain Medtronic pacemaker leads failed at abnormally high rates—about 10 percent or greater by the third year after implantation. There was much concern about the history and status of Medtronic's premarket notification (510k) submissions for polyurethane leads; major manufacturing and design changes that could affect the safety and effectiveness of their leads occurred without any FDA premarket scrutiny. Lead innovations had been designated "substantially equivalent" because leads performed the same function as the earlier products, but these were clearly very different in design, materials, and structure from their predecessors.
The FDA subsequently modified its procedures for reviewing premarket notification applications because of its experiences with Medtronic. By the mid-1980s, FDA required more evidence of comparable safety and effectiveness to support substantial
equivalence decisions: results of all types of testing, more elaborate statistical analyses of test data, and, for cardiovascular devices that are life-supporting, life-sustaining, or implanted, summaries of equivalence similar to summaries of safety and effectiveness required for premarket approval.[34]
Food and Drug Administration, Guidance on the Center for Devices and Radiological Health's Premarket Notification and Review Program (Department of Health and Human Services, 1986).
The underlying premise for 510k procedures was that a product was substantially similar to one on the market, and presumably its safety and efficacy had already been determined. It is fundamentally inconsistent to have innovative design and manufacturing changes enter the market in this fashion. After over a decade, the FDA finally began to rectify the problem.The Burdens of Class III: The Case of Extracorporeal Shock-wave Lithotripsy
Only a very small percentage of devices are placed in Class III and therefore are subject to the full premarket review similar to drug evaluations. This process can be extremely time-consuming and expensive for the producer. Of course, the purpose is to produce sufficient safety and efficacy data to ensure that the product meets the statutory standards before entering the market. The introduction of extracorporeal shock-wave lithotripsy (ESWL) in 1984 illustrates dynamic innovation in the private sector and its interrelationship with regulation.
Kidney stones in the urinary tract (urolithiasis) develop when minerals, primarily calcium and oxalate, form crystals rather than being diluted and passed out of the body. More than 300,000 patients a year (70 percent of them young to middle-aged males) develop kidney stones. For many, treatment with fluids and painkillers is sufficient; for 20 to 40 percent, the stones cause infections, impaired kidney function, or severe pain and warrant more aggressive intervention. Until the last decade, surgery to remove the stones was the only form of medical help for severe kidney stone problems.[35]
Deborah B. Citrin, "Extracorporeal Shock-wave Lithotripsy," Spectrum (Arthur D. Little Decision Resources, August 1987): 2:85-88.
The first major advance was in the early 1980s. Percutaneous endoscopic techniques permitted a physician to make a small incision and attempt stone extraction or disintegration using a special scope. The second major advance was ESWL. Its most exciting feature was that it offered a noninvasive way to treat kidney
stones. The first ESWL devices required the patient to be placed in a water bath. After X-ray monitors positioned the patient, a high-voltage underwater spark generated intense sound waves. The resultant waves disintegrated the stone into fine bits of sand that could easily pass out of the body. (The term lithotripsy comes from classical Greek and means "stone crushing."[36]
Alan N. G. Barkun and Thierry Ponchon, "Extracorporeal Biliary Lithotripsy: Review of Experimental Studies and a Clinical Update," Annals of Internal Medicine 112 (15 January 1990): 126-137, 126.
) Subsequent technological modifications eliminated the need for the water bath, and mobile units were developed. Devices that use optical fibers as conduits for laser light pulses that fragment the stones are currently in experimental stages of development.[37]Gary M. Stephenson and Greg Freiherr, "High-Tech Attack: How Lithotripters Chip Away Stones," Healthweek, 4 December 1989, 25.
While ESWL is an exciting innovation, several factors might have led to skepticism about its likely commercial success. The equipment was very expensive (early models cost at least $1.5 million), there was a viable surgical alternative, and the patient base was small and likely to remain so. And because the device was in Class III, it was subject to the highest level of premarket scrutiny.[38]
Federal payment policies were not critically important here because only a small percentage of kidney stone patients are covered by Medicare. Thus, the regulatory issues can be seen clearly.
The product took thirteen months to receive FDA approval, slightly longer than the average of one year. Despite this delay to market, it diffused rapidly once available. There were over two hundred lithotripters in operation within two years of introduction (see figure 17). The market now includes 220 devices and is basically saturated. Of ten firms in the market, only four have received FDA approval; the others have devices in investigational stages (see table 7). The market leader is Dornier Medical Systems, the first to receive a PMA; the others include Medstone International, Diasonics, Technomed International, and Northgate Research.[39]
For data on the industry, see Biomedical Business International 11 (15 July 1988): 99-101.
The next generation of machines is already in development. In a relatively short time, there have been major improvements in the original device; other designs, such as those that use laser technology, are on the horizon. There have been a number of creative marketing solutions to the problems of high cost and low patient volume. Entrepreneurs have put together joint ventures with physicians and hospitals that ensure a broad patient base, lower the unit cost of treatment, and amortize the cost of the device. Some free-standing centers have developed relationships with providers of other forms of kidney stone treatment so
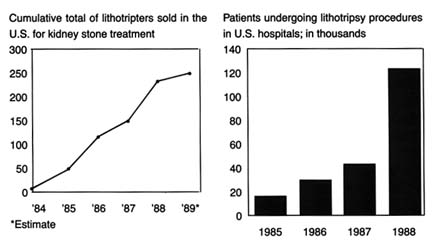
Figure 17.
Treating kidney stones with shock waves. Adapted from Ron Winslow,
"Costly Shock-wave Machines Fare Poorly on Gallstones," Wall Street
Journal , 9 February 1991, B1.
that comprehensive services and alternative treatments to lithotripsy are all available in one location.[40]
Miles Weiss and Greg Freiherr, "Romancing the Market for Stones," Healthweek, 4 December 1989, 18-20.
What lessons can we learn from this case about the nature of innovation in the device industry? How can we explain the success of this expensive, highly regulated technology? One possible explanation is that promising and truly useful technologies usually succeed despite the barriers placed in their paths. However, it may be that the dynamism and creativity are based on the expectation of enormous market expansion through the application of this technology to patients with gallstones, a much more prevalent clinical condition than kidney stones. There are 20 million gallstone patients in the United States, with 487,000 gall bladder removals in hospitals every year. Medicare plays an important role because gallstone disease affects many elderly people. The treatment of gallbladder disease is a $5 billion market. If lithotripsy could be applied to some of these patients, hospitals could avoid many of the surgical costs and the firms could compete for this greatly expanded market.[41]
Tim Brightbill, "Gallstone Lithotripsy Suffers FDA Setback," Healthweek, 4 December 1989, 25-26. For a more scientific discussion of gallstone, or biliary, lithotripsy, see Michael Sackmann et al., "Shock-wave Lithotripsy of Gallbladder Stones: The First 175 Patients," New England Journal of Medicine 318 (18 February 1988): 393-397. See also Barkun and Ponchon, "Extracorporeal Biliary."
(See figure 18.)Whether that expansion will occur is now in doubt, and here is where the policy process reenters. In October 1989, an FDA
advisory panel recommended that the agency disapprove the PMAs filed by Dornier Medical Systems and Medstone International for gallstone (biliary) lithotripters. The panel members expressed concern about the safety data in the PMAs. Questions were also raised about the effectiveness of lithotripsy for destroying all gallstones. Preliminary evaluations revealed that only a small percentage of patients with gallstones may benefit from EWSL.[42]
Brightbill, 25-26.
The delay (or possible denial) in marketing approval may allow competitors to catch up with the two leaders, although the ultimate clinical usefulness of biliary lithotripsy remains uncertain. Manufacturers have been slow to gather sufficient data because the lack of any third-party reimbursement for this new procedure has limited the number of patients who have received it. In addition, because the drugs used in conjunction with the treatment work slowly, studies are often time-consuming. In the meantime, alternative treatments are developing, including a laser that views and snips off the gallbladder and pulls it out through a small incision. Other experiments include a rotary device that whips gallstones until they liquefy and then draws out the resulting "soup."[43]
Ron Winslow, "Costly Shock-wave Machines Fare Poorly on Gallstones, Disappointing Hospitals," Wall Street Journal, 9 February 1990, B1, B6.
The failure of biliary lithotripters to receive FDA approval may only be a temporary and minor delay. It may also indicate that the technology is inappropriate for the proposed use, and the FDA is sagely valuing safety concerns over the desires of the innovative firms to rush to market. Or we may be seeing a regulatory failure in which the FDA is inappropriately obstructing a valuable innovation from the marketplace. The FDA's decision delays reimbursement from third-party payers, including Medicare, which will rarely pay for unapproved technologies, further burdening the innovators. The FDA approval does not necessarily guarantee Medicare's coverage of the procedure. The Health Care Financing Administration (HCFA), Medicare's payment authority, makes its own assessments of new technologies for coverage and payment decisions, often independently of FDA findings.[44]
For a complete discussion of the post-1983 Medicare coverage process, see chapter 7.
The lithotripsy industry remains dynamic, highly innovative, and very competitive. However, the market for kidney stone treatment is saturated and not expanding. No improved
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
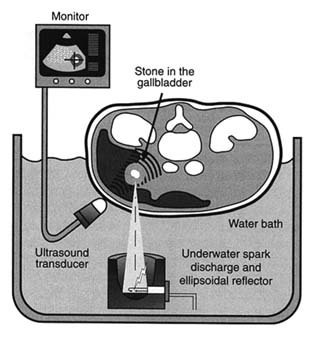
Figure 18. Electrohydraulic shock-wave lithotripter.
Source: Healthweek , 4 December 1989, 25.
technology to date has left competitors outmoded. Whether the expansion for use in gallstone treatment will occur depends upon the public sector—the FDA and Medicare—as well as private third-party payers. The layering effect becomes important here because if the FDA has not approved a treatment, then the HCFA will not cover it. And, even if the procedure has been FDA approved, approval does not ensure private or public sector third-party payment.
Postmarket Controls: Reporting Failures
The postmarket surveillance system has four main components: (1) voluntary reporting of problems from users, such as doctors or hospitals to the FDA, manufacturers, and others; (2) mandatory reporting of known problems by manufacturers to the FDA; (3) monitoring and analysis of problems by the FDA; and
(4) a recall process to correct products or remove them from the market.[45]
House Subcommittee on Health and the Environment, statement of Charles A. Bowsher, comptroller general, Medical Devices: The Public Health at Risk, 6 November 1989.
Significant controversy has surrounded the reporting requirements. Until 1984, the reporting of adverse effects associated with medical devices was voluntary. The FDA received reports from physicians, hospitals, and manufacturers, and these data were entered into the FDA's Device Experience Network (DEN). Investigations revealed that adverse reactions were seriously underreported.[46]
Senate Committee on Governmental Affairs, Report to the Chairman: Early Warning of Problems Is Hampered by Severe Underreporting (Washington, D.C.: General Accounting Office, December 1986).
The FDA promulgated a mandatory medical device reporting rule (MDR) that went into effect in December 1984. The key element in the rule is that manufacturers and importers must report to the FDA when they receive or otherwise become aware of information that reasonably suggests that a product has caused or contributed to serious injury or death, or has malfunctioned and is likely to cause harm if the malfunction recurs. There are tight time frames for reporting. In general, an injury is considered serious if it is life-threatening or results in permanent impairment of a bodily function or permanent damage to body structure. Users such as hospitals and doctors can report voluntarily, but they are not required to do so.[47]
Final Rule, 49 Federal Register 36326-36351 (14 September 1984).
Serious problems that came to light through MDR were burns related to the misuse of apnea monitors and early depletion of batteries for portable defibrillators.From the FDA's perspective, MDR also serves as a barometer of trends of adverse product performance. The FDA has received 18,000 MDR reports since the regulation became effective. Eighteen cardiovascular, anesthesiology, and general hospital devices have accounted for 70 percent of the reports.
There has been a great deal of criticism of MDR from the industry, which has argued that the system forces overreporting because of the breadth of the definitions and the short time frame.[48]
Office of Management and Budget Symposium, March 1986.
On the other hand, some health advocates maintain that the reporting system is hampered by the lack of FDA jurisdiction over hospitals and physicians, neither of which can be ordered to report malfunctions. Recent GAO studies of the FDA device recalls (removal from the market of a product that violates FDA laws) found that only half of all recalls had an MDRreport associated with them. The FDA became aware of the majority of device problems in ways other than through the required reports, and it did not have reports available in the majority of cases when decisions about health hazards were made. The GAO concluded that "this suggests that the reports have not served as an effective 'early warning' of device problems serious enough to warrant a recall."[49]
Medical Device Recalls (Washington, D.C.: General Accounting Office, August 1989).
Congress conducted further investigations into reporting failures in late 1989. At a hearing held by the House Subcommittee on Health and the Environment, Congressman Sikorski excoriated manufacturers who failed to report hazards associated with their products. Citing GAO statistics, he noted that 48 percent of high-risk products that had been recalled had never reported problems to the FDA. Grieving parents also explained that their son died because an infant monitoring system failed to notify them that he was not breathing.[50]
House Subcommittee on Health and the Environment, Committee on Energy and Commerce, statement of Gerald Sikorski and testimony of Michael B. Davis, Sr., and Cory J. Davis, 6 November 1989.
The comptroller general testified that the GAO had investigated all major components of the postmarket surveillance system since 1986. It found that the FDA was receiving more information than it previously had, but it also noted that the degree of compliance with MDR could not be established, that the FDA's data-processing system was not adequate to handle the reports it did receive, and that the results of the analyses were often not definitive.[51]
House Subcommittee on Health and the Environment, testimony of Charles Bowsher, Medical Devices, 13-15.
Thus the controversy over the FDA's ability to perform its duties under the law continued into the 1990s.Informal Powers
It is important to remember, however, that a federal agency like the FDA need not always initiate formal action to get results. Although there have been substantial problems with reporting requirements, the FDA has exercised its other postmarketing surveillance powers effectively—and often behind the scenes. The case of toxic shock syndrome illustrates this point. It is particularly striking to compare the FDA's informal power in this case to the Dalkon Shield recall in 1976, before the passage of the device law. When problems arose relating to the Dalkon Shield, the FDA had no clear authority to order a product recall.
When the toxic shock crisis arose in the early 1980s, a very different FDA, with a larger stock of potential tools, took charge.
Tampons were introduced in the 1930s and the Tampax brand dominated the market for decades. In the 1970s, Playtex, Johnson & Johnson, and Kimberly Clark marketed tampon varieties. Procter & Gamble, the large consumer product company, entered the market in late 1979. After a $75 million massive media and direct marketing campaign in late 1979, its Rely brand had acquired a 20 percent share of the billion-dollar industry.[52]
For a discussion of this case in greater detail, see Susan Bartlett Foote, "Corporate Responsibility in a Changing Legal Environment," California Management Review 26 (Spring 1984): 217-228, 221.
Toxic shock syndrome (TSS) is a rare and mysterious disease characterized by high fever, rash, nervous disorders, and potentially fatal physiologic shock. TSS was not initially associated with tampon use. In January 1980, epidemiologists from Minnesota and Wisconsin reported a total of twelve TSS cases to the federal Centers for Disease Control (CDC). The data revealed surprising patterns—all patients were women using tampons. Through that spring, the CDC received additional reports from many states. Following a retrospective case study in May 1980, tampon manufacturers were made aware of the hazards tentatively associated with their products. There were many unanswered medical questions, but because millions of women used tampons, there was fear of potential widespread injuries. In a study of fifty women who had TSS in September 1980, the CDC revealed that 71 percent of those surveyed had used the Rely brand. Procter & Gamble officials immediately defended the product. Within a week, however, they suspended sales of Rely and signed a comprehensive consent decree with the FDA.
The speed of this action can be attributed to several factors, including the specter of product liability suits and the fear of general rejection of Procter & Gamble products. The FDA played an important part in the response. It was under significant public and media pressure to protect the public from TSS, despite the unanswered medical questions and the inconclusive findings in the small CDC studies. Procter & Gamble's timing was clearly determined by the FDA, who had called a meeting one day after the release of the damaging CDC study in September. The agency gave the company one week to generate evidence that the product was safe.
Despite enormous effort, Procter & Gamble could not rebut the CDC's scientific findings in that time, and it feared that overt refusal to cooperate with the FDA would have damaged the company's reputation. The result was a voluntary withdrawal of the product. This action occurred in the shadow of the FDA's powers; the decree itself states that the FDA was "contemplating the possibility of invoking the provision of [medical device law] to compel the firm" to recall Rely.[53]
Consent decree signed by Procter & Gamble and the FDA (22 September 1980).
Assessing FDA Impact on Medical Device Innovation
The goal of FDA regulation is to establish a threshold of safety and efficacy for medical devices. The regulatory process does not intend to destroy innovation or drive away "good" technology. The challenge is to establish a balance between safety and innovation. Has the balance been achieved?
Opinions are strong on all sides. Congress has generally approached the evaluation from a consumer perspective. It has sharply and regularly criticized the FDA for underregulation and failure to enforce regulatory standards. On the other hand, industry representatives have complained about unnecessary and cumbersome regulatory burdens. Who is right?
The data are inconclusive as to the effects of regulation on innovation. Generally, however, studies have indicated that, in the aggregate, device regulation has not inhibited the introduction of new goods.[54]
U.S. Department of Health and Human Services, A Survey of Medical Device Manufacturers, prepared for the Bureau of Medical Devices, Food and Drug Administration by Louis Harris and Associates, no. 802005 (Washington, D.C., July 1982). This comprehensive, but early, survey concluded that the impacts were minor, although smaller firms might feel regulatory effects more strongly than larger ones.
Although few negative effects on equipment development have been found, small manufacturers may bear a greater burden than larger ones. Some researchers found that smaller firms were less likely to introduce Class III devices after device regulation was in place.[55]Oscar Hauptman and Edward B. Roberts, "FDA Regulation of Product Risk and the Growth of Young Biomedical Firms" (Working paper, Sloan School of Management, Massachusetts Institute of Technology, 1986).
In a study of new product introductions of diagnostic imaging devices, however, the results offered no evidence of bias against small firms.[56]Mitchell, "Dynamic Commercialization." Mitchell tried to measure regulatory effects by comparing the types of firms that introduced computed tomography (CT) scanners and nuclear magnetic resonance (NMR) imaging devices. CT diffusion preceded the 1976 law and NMR was introduced subsequently. He hypothesizes that if there were more start-up companies in computer homographics than in magnetic resonance imaging, then one could conclude that there was a regulation induced bias. His data indicates there was no evidence of small-firm liability.
The Medical Device Amendments of 1976 present a watershed for medical device innovators. The passage of the law forced all producers to consider the potential impact of federal regulation. This possible federal intervention, both as a barrier to and a manipulator of the marketplace, inextricably links the private producer to the FDA. The law attempted to deal with the
complexity and the diversity of medical devices; it is these characteristics of the industry that have led to problems of effective regulation. Even at the lowest levels of scrutiny, compliance with FDA requirements involves time and expense to the producer. For devices in Class III, the delays and the costs are much higher.
It is obvious, however, that to firms that must meet regulatory requirements, the barriers may seem high. The impact, one can conclude, is spotty across various manufacturers and types of products. Clearly the FDA has not fully implemented all the provisions of the law, and there are powerful critics of both the FDA and the industry in Congress. If the law were fully implemented or made more stringent, then greater impacts on more firms would be inevitable. The policy question that remains, then, is whether we need more safety; if so, at what cost to the producers? Can we balance the interests of innovation and safety?[57]
These issues will be pursued further in chapters 9 and 10.
Another policy designed to inhibit discovery of devices was emerging alongside the regulatory arena. Rules relating to product liability also had the potential to influence the health of our patient. It is to these legal constraints that we now turn.