Chapter 8—
The Endomembrane System and the Integration of Cellular Activities
8.1—
Introduction
The invention of ultra-thin sectioning gave electron microscopists the first sight of a new level of organization within the cell: a system of membranes forming microstructures and sub-compartments throughout the protoplasm. Some components of this system are small and discrete: the mitochondria, plastids and microbodies. Because they therefore survive cell breakage intact, biochemists have been able to characterize the specialized functions of each type. Others, the nuclear membrane, the plasma membrane (plasmalemma) and the tonoplast, bound discrete compartments so large that, though they do not survive cell breakage intact, many of their specialized functions have been worked out by cytologists and physiologists using whole cells.
Subtract these organelles from the membrane system of the cell and the remainder, a considerable bulk of material, is arranged as parallel, paired sheets fused at the edges to enclose a narrow and empty-looking lumen. The membranes are also fused to each other around holes in the twin sheet, and these fenestrations are occasionally so frequent as to reduce the paired membrane to a layer of tubules, or even isolated vesicles. A small proportion of this doublemembrane system, the Golgi apparatus, is organized into units of recognizable morphology, the Golgi bodies. The rest, the endoplasmic reticulum (ER), is relatively amorphous and, more often than not, distributed apparently haphazardly about the cytoplasm. Offering few clues for the microscopist and a host of problems for the biochemist, this material and its function in the cell is only just beginning to be understood.
8.2—
Techniques
8.2.1—
Electron Microscopy
Thin (ca 50 nm) sections provide a very limited image of the membrane system. Seen in profile, not much can be told about the way its parts are, or are not, connected. What is described from an isolated thin section as a collection of vesicles may in fact be a network of tubules, anastomosing in three dimensions, or a stack of paired lamellae, extensively fenestrated, or even a single tubule, coiled back on itself many times (Fig. 8.1). There are methods which provide information on structure in three dimensions. Thanks to the tremendous depth
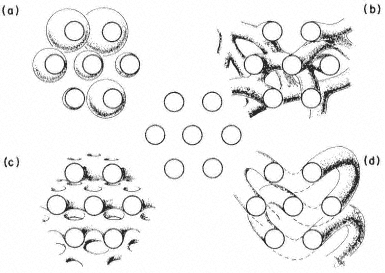
Figure 8.1
Identical thin sections can be produced from quite different structures:
(a) a collection of vesicles
(b) a three-dimensional network of tubules
(c) a stack of fenestrated lamellae
(d) a single, twisted tubule
of focus of the electron-microscope, using high-voltage electrons it is possible to view all the organelles within a thick (2 µm) section. The specimen can then be tilted slightly to provide stereo-pairs of micrographs.
The freeze-etch technique often provides information on membrane topography. In frozen tissues the hydrophobic bonds holding the two halves of each membrane together are virtually non-existent. The activity of cell water is so low, the lipids of the two halves of the bimolecular leaflet are no longer forced together. The freeze-fracture, which follows planes of weakness, may run through large areas of interconnected membrane. Freeze-etch studies often allow a direct comparison to be made between views of the internal surface and cross-fracture of the same membrane system, as in Fig. 8.2 where the ER is seen as continuous sheets arranged in concentric layers. Fenestrated ER can only be shown by freeze fracture (Fineran, 1973b).
To obtain a truly comprehensive picture of membrane interconnections requires serial sectioning. So lengthy and difficult it is rarely attempted, this technique has nevertheless proved highly rewarding. Not until 1973 was it discovered by serial sectioning that there is only one, highly branched mitochondrion in a yeast cell, and the same is true for the unicellular green alga, Chlorella (Atkinson et al., 1974; Gunning & Steer, 1975). Though freeze-etch pictures of plant cells have already provided much information on the shape of the endomembrane complex, thick section stereomicrography and serial sectioning are as yet techniques of the future.
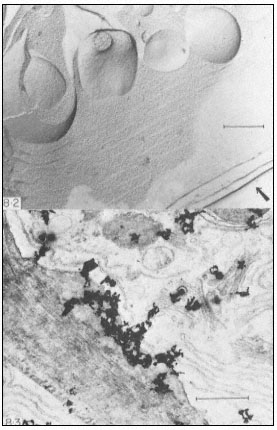
Figure 8.2
Freeze-etch preparation of pea root tip. ER is seen in cross-fracture and surface view,
and forms almost continuous sheets arranged in concentric layers. The arrow indicates
the direction from which heavy metal was evaporated and the scale line represents 1 µm.
(From Northcote, 1968.)
Figure 8.3
Autoradiograph of a thin section of a wheat root cap cell exposed to [6-3 H] glucose
for 30 minutes. Silver grains are located over slime- polysaccharide deposited between
the wall and the plasmalemma, and over Golgi bodies in the cytoplasm. The slime
deposit is sited under a gap in the peripheral ER. The scale line represents 1 µm.
(From Northcote & Pickett-Heaps, 1966.)
High resolution autoradiography is a powerful tool in the investigation of the synthetic activities of endomembranes. First, the living tissue is given a pulse of tritium-labelled precursor. At fixed intervals of time afterwards, portions of tissue are prepared for electron-microscopy, sectioned and the stained sections are coated with a very thin layer of radiosensitive, silver halide emulsion. To prevent the development of this emulsion by chemicals in the section, an inert layer of carbon is deposited on the sections before they are coated. From
the changing distribution of silver grains over the organelles of the cell, the pulse of radioactive precursor can be followed from the site of synthesis of the first insoluble product to the final resting place of the completed polymer (Fig. 8.3). Because the b -particle may have moved in the plane of the section before hitting the emulsion, the practical limit of resolution of the technique is 100 nm and since the organelles in the cell are fairly crowded it may not be possible to decide unequivocally which of them contained the labelled material.
Proper controls for autoradiographic experiments would have to include some method of identifying the radioactive product in the section. For example, pre-treating the section with solutions of degradative enzymes specific for the polymer in question should eliminate the radioactivity associated with the organelles involved in its synthesis. Also, a precursor specific for the product under investigation should be used, e.g. [3 H]-orotic acid for RNA. Nonspecific precursor, such as [3 H]-glucose, is only useful with cells producing a single polymer-type, e.g. slime polysaccharide in root-cap cells or cellulose in differentiating xylem vessels. Always important, but especially necessary in this case, is a back-up biochemical analysis showing the distribution of label from the precursor in all polymeric fractions at the times that the tissue portions were fixed for electron-microscopy. It must be admitted that proper controls of this nature are hardly ever reported.
8.2.2—
Biochemistry
To isolate the various parts of the endomembrane complex, the biochemist breaks open the cells of a lump of tissue, in a pH-buffered solution to neutralize vacuolar acids, and separates membrane fractions from the rest of the cytoplasm by centrifuging. To homogenize is to use high shear forces to fragment all membranes. The little pieces spontaneously re-anneal forming small vesicles or 'microsomes'. Small quantities are homogenized in some sort of pestle and mortar arrangement, large quantities in a blender. Any compartmentalization of soluble substances in organelles is lost by this method, and the in/out orientation of the membranes of the vesicles does not necessarily represent that in vivo.
To prepare intact organelles, shear forces are minimized by crushing or chopping the tissue in a solution which is also buffered osmotically to prevent bursting. A few organelles will inevitably be broken and many of them will be left behind in pieces of more or less intact tissue. So, unlike homogenization, quantitative recovery of membrane from the tissue does not occur and thus it is impossible to relate directly, say, enzyme activities in isolated organelles, to the total activity in the tissue.
Differential centrifugation involves centrifuging at successively higher speeds for longer times to effect a separation on the basis of size. Thus debris and cell wall are spun out at 4,000 g for about 15–20 minutes, organelles at 10,000 g for about 30 minutes, and total membrane by 100,000 g for 1 hour. The membrane
which pellets between 10,000 g and 100,000 g is referred to as a microsomal fraction and this material is often assumed to be ER, which probably does make up the bulk of it. Differential centrifugation is now only used to prepare organelle or microsomal fractions for subsequent analysis by density gradient centrifugation or, since resuspension of pelleted organelles causes some breakage, to remove debris before the total membrane material plus soluble cytoplasm is layered onto a gradient (Lord et al., 1973).
Density gradients are prepared in the centrifuge tube by pouring in a progressively less concentrated solution of sucrose in the buffered medium. The membrane preparation is added on top and centrifuged into the gradient. In an isopycnic separation, centrifugation is continued until the components of the sample reach their equilibrium positions in the gradient and so separation is effected on the basis of density alone. In a rate zonal separation, centrifugation is stopped before equilibrium is reached. Smaller particles sediment faster and so the ordering of components in the gradient is determined by size as well as density. Continuous gradients in which the density changes linearly with distance over a range selected from 1.06 g cm–3 (16% w/w sucrose) to 1.23 g cm–3 (50% w/w sucrose) are conventionally used to separate the components of membrane fractions. When the density of a particular component is known, discontinuous or 'step' gradients are sometimes used for bulk preparation. However this method does not produce fractions as pure as those from continuous gradients. A heavy particle arriving late at an already crowded interface between two densities in a step gradient may be trapped there or, as appears to happen more often, plummet through carrying light membranes down with it. (Note that results from sucrose gradients cannot be compared with those from experiments involving gradients of 'ficoll', a synthetic sucrose polymer sometimes used, since sucrose is osmotically active and organelles and vesicles lose water progressively as they fall through a gradient of it.) Centrifuging organelles and microsomes through linear density gradients produces distinctive patterns in the distribution of protein in the gradient, so it seems the endomembrane complex can be resolved into different types of membrane. The next step is to identify them.
If the gradient fractions are fixed and embedded for electronmicroscopy, stained thin sections can reveal the presence of organelles with a characteristic morphology, such as attached ribosomes. It would involve a lot of work to establish that a particular organelle was not present. Negative-staining is easier and faster, but less reliable since phosphotungstic acid solution is a powerful protein extractant and so, unless the membranes are glutaraldehyde-fixed, alters the appearance of organelles (Mollenhauer et al., 1973). Many membrane fractions, after conventional post-fixation with osmium and uranyl acetate staining of sections, appear as simple vesicles with no recognizable morphology. Efforts have been made to develop electron-dense stains which are specific for individual membranes of the cell, but so far only the plasmalemma has been stained selectively with any success using periodic acid and various heavy metal
anions, e.g. chromate and phosphotungstate (Leonard & Van Der Woude, 1976). However, the chemical basis of the staining reaction is by no means certain (it is likely that the carbohydrate moiety of plasmalemma glycoproteins is involved) and its specificity for this membrane cannot always be repeated.
Gradient fractions can be characterized by various enzyme activities associated with them. The enzyme proteins may be freely soluble and enclosed by the membrane, or loosely associated with the membrane surface (extrinsic), or firmly bound in the membrane (intrinsic). Obviously only intrinsic enzymes can be trusted as markers. For organelles with unique and well-characterized functions, enzymes provide reliable identification, e.g. succinate dehydrogenase, an enzyme unique to the tricarboxylic acid cycle and very firmly bound, is used to indicate the presence of inner mitochondrial membrane. Otherwise, to determine the distribution in the cell of an enzyme activity localized in a gradient is a major problem.
Sometimes enzymes can be located in thin sections of cells by cytochemical techniques for electron-microscopy, e.g. specific phosphatases can be tracked down if inorganic phosphate released from supplied substrate is precipitated in situ on thin sections by heavy metal cations. Thiamin pyrophosphatase (TPPase) was shown to be a marker for the Golgi body in vertebrate cells in this way. The specificity of the reaction must be checked by comparison of results obtained using a range of alternative substrates. An enzyme localized by cytochemistry on thin sections cannot with certainty be used to establish the identity of membrane fractions which have not been treated with glutaraldehyde or heavy metal cations, both of which are powerful inhibitors of many enzymes.
An association of enzymes with particular membranes of the cell has sometimes been built up as a result of some knowledge of cell physiology, e.g. Na+ /K+ stimulated ATPase and callose synthetase are thought to be markers for plasmalemma since salt uptake and callose synthesis take place at the cell surface. However, the limitation of callose synthesis to the outer surface of the plasmalemma in vivo may be due to substrate availability and it would seem equally likely that the tonoplast, for which at present there is no marker enzyme, is also involved in salt uptake.
Alternatively, enzyme markers become accepted by long association. Thus, because the activity of NADH-specific cytochrome c reductase correlates well with the proportion of ribosome-bearing fragments in membrane fractions, this enzyme is widely accepted as a marker for ER. (Note that the inner mitochondrial membrane also shows NADH-specific cytochrome c reductase, which differs from that of the ER in being sensitive to antimycin A. mitochondria must be shown to be absent from the fraction and the activity demonstrably antimycin-insensitive when this marker is used for ER.) This sort of association between an enzyme and a specific membrane is only of any use for tissues and organisms in which it has been established as there is no reason to believe that markers are universal. Thus, glucose-6-phosphatase is an ER-marker in certain cells only, e.g. vertebrate liver and kidney, and together with 5'-adenosine
monophosphatase (AMPase, 5'nucleotidase), a marker for plasmalemma in animals, does not occur in the equivalent membranes of plant cells.
Note that while the detection of marker enzyme activity can indicate which types of membrane are present, the absence of activity may be due to failure of the assay for a number of reasons other than absence of the enzyme. Also, no membrane fraction can be established as 'pure' on the basis of marker enzymes while there are still types, like the tonoplast, for which a marker does not exist.
8.3—
Composition and Characteristics of the Membrane Types
There is considerable evidence for broad homology between the nuclear membrane and the membranes of the ER, and some indications that the outer mitochondrial membrane and the plastid envelope, while essentially similar to the ER, are intermediate in composition between it and the specialized inner membranes of these organelles. From the little that is known of their composition, the membranes of the Golgi apparatus are closely allied to those of the ER, but the plasmalemma is markedly different from this membrane.
8.3.1—
Nuclear Membrane
Nuclear membrane is prepared from isolated nuclei by sonication to fragment the membrane, high salt treatment to remove associated nucleic acid, and density gradient centrifugation to collect the fragments. In a careful study, taking precautions to minimize phospholipid loss by enzyme degradation during isolation, Philipp et al. (1976) found no difference between the lipid and fatty acid composition and the nature and level of enzyme activities in nuclear membrane and ER from plants. Both membranes, for example, had NADH and NADPH-specific cytochrome c reductases at similar levels, and both possessed a cytochrome of the b5 type. They could be distinguished by the higher mean buoyant density of nuclear membrane (1.20 g cm–3 ), attributed to a higher content of nucleic acid and protein. (Values between 1.08 and 1.12 g cm–3 are recorded for the density of ER without attached ribosomes in sucrose gradients, and between 1.16 and 1.18 for the mean buoyant density of 'rough' ER.) In animal cells ER and nuclear membrane appear to be identical except for the higher density of the latter (Franke, 1974). They also resembled their animal counterparts in having a higher proportion (60%) of their phospholipid as phosphatidyl choline ('lecithin') than the other membranes of the cell. Phosphatidyl ethanolamine was the second most abundant phospholipid at 20–24%. They differed from animal nuclear membrane and ER, and resembled other plant membranes in their high sterol content (ca 30%), and high content of unsaturated fatty acids as esters in the lipid. Oleate (18:1) and linoleate (18:2) accounted for 80% of these and palmitate (16:0) made up most of the rest.
In thin sections, the outer nuclear membrane is often seen to bear ribosomes on its cytoplasmic face, just like both membranes of the ER, and when nuclei were isolated from pea shoots in high concentrations of Mg2+ , the outer membrane was densely covered with ribosomes (Stavy et al., 1973).
On the basis of lipid composition, enzymic activities and ability to bind ribosomes, nuclear membrane cannot be distinguished from ER. It differs morphologically in possessing nuclear pore complexes which probably contribute to its higher buoyant density.
8.3.2—
Outer Mitochondrial Membrane
The outer membrane can be released from isolated mitochondria either by a slightly hypotonic medium (osmotic lysis) or digitonin to which the inner membrane is relatively resistant. The heavy, intact inner membrane vesicles and their enzymic contents are then easily separated from the light, fragments of outer membrane.
Turnip outer mitochondrial membrane was found to have NADH-specific cytochrome c reductase and a b5 -type cytochrome, just like the microsomal fraction, from which it differed in lacking firmly bound, NADPH-specific cytochrome c reductase (Day & Wiskich, 1975). Compared with the bulk microsomes, the inner membrane of cauliflower mitochondria had a low (40 %) proportion of its phospholipid as phosphatidyl choline due to replacement by the specialized mitochondrial phospholipid, diphosphatidyl glycerol ('cardiolipin'). The outer membrane resembled the inner membrane in that only 40% of its phospholipid is phosphatidyl choline, but was intermediate between this membrane and the microsomes in its content of diphosphatidyl glycerol (Moreau et al., 1974). Isolated plastids and mitochondria have been shown to contain less of the plant sterols sitosterol and stigmasterol than the other membranes, but about the same concentration of cholesterol in µg mg–1 protein. As a result, the sterol content of these organelles is only one-tenth that of the bulk membranes of the cell (Hartmann et al., 1973). Mannella & Bonner (1975) estimate the ratio of sterol:phospholipid in the outer mitochondrial membrane as 0.17 for mung bean hypocotyl and 0.06 for potato tuber. These authors report that the degree of unsaturation of the fatty acids in outer membrane lipids was intermediate between that of light microsomes and the value for the highly unsaturated lipids of the inner membrane. For mung bean hypocotyl, the average number of double bonds per fatty acid was 1.3 for light microsomes, 1.7 for the outer and 2.0 for the inner membrane. The same pattern was seen in experiments on potato tuber, although in cauliflower Moreau et al. (1974) reported that the outer membrane was more saturated than the microsomal membranes.
In summary, the plant outer mitochondrial membrane has enzyme activity which shows it to be ER-like, but in its lipid composition it has characteristics intermediate between the ER and the very specialized inner membrane.
8.3.3—
Chloroplast Envelope
This membrane is prepared from isolated chloroplasts by osmotic rupture. Chloroplast lamellae are chiefly composed of galactolipid (92% of the lipid), especially monogalactosyl diglyceride and the low content of phospholipid is largely made up of phosphatidyl glycerol. The fatty acids are highly unsaturated, with linolenate (18:3) as the major type at 83 mole %. Trans-D3 -hexadecenoate is a minor fatty acid component unique to the lamellae. The envelope was found to differ from the microsomes and resembled the lamellae in its high galactolipid content, in the complete absence of phosphatidyl ethanolamine, and relatively high proportion of linolenate (18:3). It also shared a number of features with the microsomal fraction in respect of which the envelope differed from the lamellae. These were: phosphatidyl choline as major phospholipid, digalactosyl diglyceride as the major galactolipid, and a relatively high proportion of palmitate (16:0) amongst the esterified fatty acids (Mackender & Leech, 1974).
Like the outer mitochondrial membrane, the chloroplast envelope appears to be a curious chimaera, intermediate between the ER and the specialized membrane system it encloses.
8.3.4—
Golgi Membranes
Membrane fractions enriched in Golgi bodies are prepared by density-gradient centrifugation of organelles obtained after chopping plant tissues. Glutaraldehyde is routinely added to the extraction medium to 'stabilize' the structure of the Golgi bodies though it now seems that the organelle survives preparation intact and only requires this fixative for electron microscopy (Bowles & Kauss, 1976). Golgi bodies from plant sources sediment with the fraction of buoyant density 1.12 to 1.15 g cm–3 in sucrose gradients and are identified in gradient fractions by their distinctive morphology. Thin section studies of whole cells in higher plants show the Golgi body as a stack of 5 to 8 closed membrane sacks. Each sack, or 'cisterna', is flattened to a disc, 0.5 to 1 m m in diameter. Around the perimeter of the disc there is progressive fenestration through, and localized swelling of, the cisternal membranes, grading into isolated vesicles beyond the edge of the disc (Fig. 8.4). In some cases a single layer of parallel fibrils has been seen in the 10–15 nm wide, electronlucent region between the cisternae. Freeze-etch pictures (Fineran, 1973a) confirm this structure for the organelle. Electron-microscope sections and negative-stain images of glutaraldehyde-fixed material show that this structure is maintained in isolated organelles. The cisternae remain joined in a stack as they are in the cell, but separate in media of high ionic strength (Mollenhauer et al., 1973).
Inosine diphosphatase (IDPase) has been used as a marker for Golgi bodies and, for some plant tissues, a single peak of activity is observed on sucrose gradients, coinciding with the Golgi body-rich fraction. However, IDPase activity
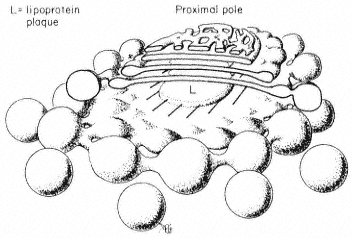
Figure 8.4
The structure of the Golgi body. Based on the
freeze-etch pictures of Fineran (1973a) which
are probably close to the real shape of this
organelle in the cell. Conventional electron
microscope methods involving fixation and
dehydration are more likely to cause distortion.
was found in purified nuclear membrane (Philipp et al., 1976), and using mung bean hypocotyl a second peak of activity was observed in light membrane characterized as ER (Bowles & Kauss, 1976).
Thin sections of Golgi body fractions from sucrose gradients show that a variety of less organized vesicles is also present, and the possibility that these preparations are contaminated with other membrane types cannot be excluded. The purest Golgi body fractions to date have been obtained from rat liver. Giving a rat ethanol induces the liver Golgi bodies to fill up with light lipoprotein particles which cause a distinctive change in the buoyant density of the organelle and serve as internal structural markers for electron-microscopy. The purified Golgi body membranes contained the cytochrome b5 electron-transport chain (NADH-specific cytochrome c reductase and cytochrome b5 ) characteristic of ER, but lacked NADPH-specific cytochrome c reductase and some other ER markers. UDP galactose:N-acetyl-glucosamine galactosyltransferase was restricted to the Golgi body fractions (Bergeron et al., 1973). Other evidence from animal cells indicates that fucosyl and sialyl transferases are primarily associated with the Golgi apparatus. 'Pure' Golgi fractions have not yet been prepared from plant material, but it has been shown that maximum activities of a UDP galactose:N-acetyl-glucosamine galactosyltransferase involved in glycoprotein biosynthesis (Powell & Brew, 1974), and UDP glucose:sterol glucosyltransferase (Bowles et al., 1976) were found in the Golgi body-enriched fraction of sucrose density gradients. The evidence then, such as it is, suggests that Golgi membranes have some affinity with the ER, but are specially enriched in glycosyltransferases involved in the synthesis of glycoprotein and glycolipid.
8.3.5—
Plasma Membrane
Plasmalemma is obtained from tissue homogenates by sucrose density gradient centrifugation after removing mitochondria whose density range overlaps that of this membrane. Reported mean buoyant densities range from 1.16 to 1.18 g cm–3 . This fraction has been identified as plasmalemma on the basis of specific staining, and characterized by the presence of Na+ /K+ stimulated ATPase activity, and glucan synthetase activity at high UDP glucose concentrations (callose synthetase), and the absence of ER and Golgi body markers (NADH-specific cytochrome c reductase and IDPase) (Leonard & Van Der Woude, 1976). Since none of the markers can be considered exclusive to the plasmalemma and non-staining membrane is always seen in the fraction, firm characterization of this membrane in plants will depend on better means of separation or identification. Investigators have tried to label the external surface of this membrane in intact cells, e.g. using radioactive iodine, hoping to trace the membrane in a gradient by its label. The most successful attempt has involved the carbohydrate-binding protein, concanavalin A (conA), which has been used to prepare plasmalemma from wall-less mutants of Neurospora and yeast protoplasts. Treatments with conA just before lysis stabilizes the membrane as large sheets and in this form it is easily separated from small vesicles. Removing the conA causes the sheets to vesiculate and now the plasmalemma can be separated from large contaminants which had co-sedimented with it (Scarborough, 1975).
Plasmalemma fractions from animal cells have a much higher content of glycoprotein and glycolipid than the other membranes of the cell and the carbohydrate moieties of these compounds form a glycocalyx over the external surface. Scarborough's work with conA suggests a similar layer is present on the surface of the fungal plasmalemma, and the specific staining of the plant plasmalemma after treatment with periodic acid, which creates reactive aldehyde groups on carbohydrate residues, implies that this is also true for the plant plasmalemma. When thin sections of protoplasts in plasmolysed onion tissue were stained for carbohydrate, only the outer surface of the plasmalemma reacted. Since this response was unaffected by treatment with concentrated cellulase and pectinase, it is unlikely that it was due to wall polysaccharide (Roland & Vian, 1971).
Bailey and Northcote (1976) have obtained a pure preparation of plasmalemma from the green alga, Hydrodictyon, by syringing out the contents of these huge cells, leaving the plasmalemma firmly attached to the wall. Since there was insufficient material for direct chemical analysis, the composition of the membrane was estimated from the radioactivity in each phospholipid after feeding [32 Pi ] to algae for 20 days, sufficient to saturate the membranes with label. Phosphatidyl choline was very much the major phospholipid, accounting for 58% of the radioactivity in identified phospholipids. Phosphatidyl serine, though only traces were found in whole cells, was a substantial component of the plasmalemma at 8%. Sterol:phospholipid molar ratios between 1.0 and 1.2
have been recorded for plant membranes isolated on a density gradient and identified as plasmalemma-rich by staining. This represents a considerable enrichment in sterol over the total (0.4) and light membrane (0.2) fractions (Hodges et al., 1972). High sterol content will tend to reduce the fluidity of the membrane and so stabilize the cell boundary. In animal cells also phosphatidyl serine is predominantly associated with the plasmalemma and, to a lesser extent, the Golgi apparatus. Again, in animal cells, there is an increase in the proportion of sterols from ER to Golgi apparatus to plasmalemma, but they differ from Hydrodictyon in that this sequence is associated with a marked decline in the contribution of phosphatidyl choline to the phospholipids.
In summary, plasmalemma-enriched membrane fractions have a set of enzyme activities very different from that of the ER. There is some evidence that this membrane, like its animal counterpart, is more glycosylated than other membranes, from which it also differs in lipid composition.
8.4—
Functional Relationships between Membranes
8.4.1—
Membrane Synthesis
Since mitochondria and chloroplasts appear to make only a small fraction of their protein and lipid, and the plasmalemma none at all, there seems little doubt that most of this material is ultimately derived from the ER which has been shown to be the only site in the cell competent to synthesize the major membrane constituents.
Microsomal fractions from onion stem and spinach leaves synthesized phosphatidyl choline from CDPcholine and phosphatidic acid and phosphatidyl ethanolamine from CDPethanolamine and phosphatidic acid (Marshall & Kates, 1973). Beevers' group have shown that the terminal enzymes for the synthesis of phosphatidyl choline, phosphatidyl serine and phosphatidyl inositol are found only in a light membrane fraction (buoyant density 1.12 g cm–3 ) prepared from chopped endosperm of germinating castor beans (Beevers, 1975). The whole cytoplasm, soluble enzymes as well as membranes, was centrifuged into a linear gradient containing 1mM EDTA. Electron-microscope sections showed that the membranes carried no ribosomes and they had sedimented separately. In gradients containing 3 mM Mg2+ , activity at 1.12 g cm– 3 largely disappeared and the membrane, NADPH-specific cytochrome c reductase, and phosphorylcholine glyceride transferase (the terminal enzyme in phosphatidyl choline synthesis) activities were all spread around 1.16 g cm–3 . This membrane was now coated with ribosomes. It was concluded that the final steps in the synthesis of these phospholipids are located exclusively in the ER (Lord et al., 1973). A key enzyme of sterol biosynthesis, transfarnesyl pyrophosphate-squalene synthetase, is almost all associated with fractions rich in ER (Hartmann et al., 1973).
Polyribosomes, the groups of ribosomes linked by mRNA and active in protein synthesis, occur free in the cytoplasm and attached to the surface of the
ER and outer nuclear membrane. They can be seen in glancing sections of these membranes as folded or spiral chains of ribosomes. The potential of the ER to synthesize membrane proteins is therefore not in doubt. There is good evidence from mammalian cells that secretory proteins are synthesized by ribosomes attached to ER and pass rapidly through this membrane into the lumen (Jamieson, 1975). Liver cells synthesize protein mainly for secretion and 70 % of the polysomes are bound. Puromycin released most of the ribosomes from rough ER isolated from liver, indicating that it is the attachment of the growing peptide to the membrane which holds the ribosomes on. Now, this does not mean that proteins synthesized by rough ER are inevitably incorporated into or through the membrane. In non-secretory animal tissue-culture cells the ribosomes were bound to ER only by their attachment to mRNA. Interestingly, for rough ER from bean hypocotyls, 80% of the bound ribosomes required puromycin for release and only 10% were released by RNase alone (the others could be removed by high salt) (Dobberstein et al., 1974). Since only a few plant cell types secrete large amounts of protein, e.g. cells of the aleurone layer of cereal grains, it appears that most of the protein made by the rough ER is being incorporated into the membrane rather than through it. However, only 20 to 25% of the cytoplasmic ribosomes were recovered in the rough ER fraction isolated from bean hypocotyls (cf liver cells) and it is quite possible that membrane proteins are made on free polyribosomes and subsequently incorporated into the appropriate membrane.
To find out more about the synthesis and movement of membrane in the cell, a number of investigators have fed radioactive precursors of lipid and protein and traced the pattern of distribution of labelled product in various cell fractions.
In rat liver cells, nuclear membrane and ER incorporate a variety of [14 C]-amino acids into membrane protein with identical kinetics, and the labelling of the phosphatidyl choline of these membranes by [14 C]-choline rules out either synthesis in the nuclear membrane and transfer to the ER, or synthesis in the ER and transfer to the nuclear membrane (Franke, 1974). These results confirm the close similarity of these two membranes. No comparable findings have been reported for plants.
When [14 C]-choline was supplied to castor bean endosperm, phosphatidyl choline in the ER was labelled before that of any other membrane and part of this labelled phosphatidyl choline appears in mitochondrial membrane within a short time (Beevers, 1975). As the enzyme studies suggested, mitochondria are dependent on the ER to synthesize the bulk phospholipids of their membranes. In similar experiments with animal tissues, the specific activity of phosphatidyl choline and phosphatidyl ethanolamine in the outer membrane was intermediate between that of the ER and that of the inner membrane. This is consistent with the movement of these phospholipids from the ER to the outer membrane to the inner membrane (not a very startling idea since access to the inner membrane from outside the mitochondrion must be via the outer). The
dependency of this organelle on the ER for phospholipids is not total. The terminal enzyme for the synthesis of phosphatidyl glycerol was found in both ER and mitochondria of castor bean endosperm. This phospholipid is a precursor for diphosphatidyl clycerol (DPG), in which the inner membrane is especially rich, and isolated animal mitochondria have been shown to synthesize phosphatidyl glycerol and DPG.
Whether the lipids of the chloroplast lamellae are made 'on site', or imported from somewhere else in the cell, is still a matter of controversy (Morré, 1975).
Another mystery is the site of synthesis of phospholipid precursors. Although fatty acid synthetase is soluble, recent work has indicated that all the activity is membrane-enclosed in the cell (Weaire & Kekwick, 1975; Donaldson, 1976), and at least part of it was found in 'heavy' organelles, proplastids and mitochondria. Certainly, when [14 C]-acetate was supplied to castor bean endosperm, lipid in organelles was labelled at the earliest measurement (5 minutes) making it unlikely that fatty acid synthesis was restricted to the ER. If mitochondria and plastids are able to synthesize some of their own fatty acids, this could explain how they maintain a high level of unsaturation against the influx of lipid from the ER, since phospholipid molecules in membranes are dynamic structures in which acyl moieties can be replaced or exchanged. In addition it is possible that the fatty acids of ER-derived phospholipid could be desaturated in the plastid or mitochondrion. Desaturases are always intimately associated with membrane systems since the participation of a special electron transport chain is required, and phospholipid appears to be the preferred substrate for the production of linoleate (18:2) from oleate (18:1) in Chlorella (Sedgwick, 1972).
Independent, internal synthesis of special components, e.g. phosphatidyl glycerol and unsaturated fatty acids, is one way the distinctive composition of organelle membranes may be maintained. Another way would be to use the intermembrane space, e.g. between inner and outer mitochondrial membranes, to control the movement of individual lipid types. The differences in lipid composition between these two membranes support the idea and there are reports of 'exchange' proteins in the soluble cytoplasm which can ferry specific phospholipids in solution between membranes (McMurray & Dawson, 1969; Morré, 1975). The fact that the ER and the outer membrane differ in composition suggests that lipid must also be 'filtered' as it passes between these two membranes. For this reason, if no other, it is unlikely that the ER is structurally continuous with these outer membranes in the cell. Although movement between the two halves of the bimolecular leaflet is restricted (see chapter 2), lipids and many membrane proteins are freely mobile in the plane of the membrane and any differences in composition would soon disappear. Conventionally prepared, electron microscope sections have provided occasional observations of interconnections between all the membrane types of the cell, but phenomena seen only infrequently by electron microscopy should be treated with suspicion. Membrane conformations are stabilized by weak bonds, and the covalent cross-linking of proteins by glutaraldehyde could force lipidic structures to
re-organize to create continuities, for example where two membranes were closely appressed. For certain species, e.g. the fern Pteris, an unusually large number of conjoinings between membranous organelles has been reported (Crotty & Ledbetter, 1973) which makes it unlikely that these observations have universal significance. The close similarity, biochemically, between nuclear membrane and ER would support observations that these two membranes are in clear luminal continuity, but can also be explained as the result of the total re-structuring of the nuclear membrane from ER at every mitosis. It has been suggested that membrane could be transferred from the ER to the outer organelle membranes by intermittent direct connections, and lipid components might also move in solution on 'carrier' proteins (Morré, 1975).
From thin section studies, electron microscopists suggested that the ER and the plasmalemma are linked functionally via the Golgi apparatus, the cell's complement of Golgi bodies. Golgi bodies can be seen adjacent to a sheet of ER or the outer nuclear membrane, aligned with the cisternae parallel to the plane of these membranes. The portion of membrane apposed to the Golgi body bears no ribosomes but projects as small tubules into the cytoplasm between. It is hypothesized that the tubules vesiculate and the 'primary' vesicles formed then re-fuse to form a new Golgi body cisterna (Morré & Mollenhauer, 1974; Gunning & Steer, 1975). Also, circular membrane profiles, similar to those in the vicinity of the vesiculated rim of the cisternae, are found close by and apparently fused with the plasmalemma. The membrane of these 'secretory' vesicles is stained heavily after treatment of sections with barium permanganate and the plasmalemma stains in the same way. ER does not stain heavily. The membranes of the Golgi cisternae show a progressive increase in staining intensity from ER-like at one pole to plasmalemma/'secretory' vesicle-like at the other (Grove et al., 1968). There is, then, ultrastructural evidence to suggest that the Golgi apparatus is linked to both ER and plasmalemma by alternating membrane vesiculation and fusion, and some aspect(s) of the transition between ER and plasmalemma membranes takes place within the Golgi apparatus. Of course, equivalent fiffinity for heavy metal is no real indication of biochemical similarity, no more than the intermediate density of the Golgi body can be considered as evidence for its function as an intermediate between ER and plasmalemma.
After feeding labelled precursors, the specific radioactivity of phospholipid in membrane fractions was reported to be highest in ER, intermediate in Golgi bodies and lowest in plasmalemma (Morré, 1975), a pattern consistent with the proposed function of the Golgi body.
Studies of the incorporation of a pulse of radioactive amino acid, [guanido-14 C]-arginine, into membrane protein in rat liver have shown that membrane protein in a 'rough microsomal' fraction was labelled very early on. The kinetics of labelling suggested that some membrane protein passed from ER to Golgi apparatus to plasmalemma. Thus, the rough ER fraction reached maximum specific activity within 10 minutes of feeding. By 20 minutes the
specific activity had fallen by one-fifth, as if part of the newly labelled protein had left the fraction. At the same time the specific activity of a Golgi bodyenriched fraction, already higher than that of the ER, continued to rise reaching a maximum at 30 minutes and declining until 60 minutes to five-sixths the maximum value. Now, between 10 and 60 minutes, the membrane protein of a plasmalemma fraction accumulated label in two phases, firstly as label was lost from the rough ER and then as label disappeared from the Golgi fraction (Fig. 8.5, Franke et al., 1971). These results indicate that:
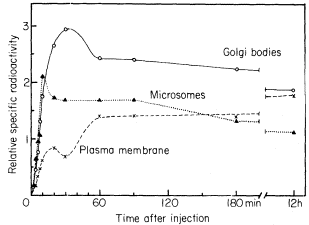
Figure 8.5
Changes in the specific activity of membrane proteins in organelle
fractions from rat liver after feeding a pulse of [guanido-14 C] arginine.

× - - - - × plasmalemma-enriched fraction (from Franke et al., 1971.)
(a) proteins of the ER may contribute to the plasmalemma without the mediation of the Golgi apparatus,
(b) only a part of the newly labelled ER and Golgi apparatus membrane protein turns over sufficiently rapidly to be able to give rise to the observed increase in plasmalemma labelling,
(c) if the newly labelled material of the Golgi apparatus is derived from the ER (and this is by no means obvious from the data since there is no lag between incorporation into ER and into Golgi apparatus fractions) then it must be transferred from a region of synthesis and rapid turnover within the bulk ER since the specific activity of the Golgi apparatus was higher than that of the ER fraction.
Similar experiments have been performed with onion stem tissue, feeding [U-14 C]-leucine (Morré & Van Der Woude, 1974). In this case a lag of 30 to 60 minutes was observed after incorporation into microsomal and nuclear membranes had begun and before Golgi apparatus and plasmalemma fractions
began to be labelled. The authors discern a second phase of incorporation into plasmalemma, lagging 30 minutes behind the initial simultaneous labelling of Golgi apparatus and plasmalemma. Their results are consistent with transfer of newly synthesized protein from microsomal to Golgi membranes in plant cells and, as in rat liver, the ER may contribute material directly as well as via the Golgi apparatus. Morré and Van Woude assumed the label in membrane fractions represented incorporation into membrane protein only. Though the cells of onion stem do not secrete protein on the same scale as rat liver, this assumption is not entirely justified. A hydroxyproline-rich glycoprotein is a substantial component of cell walls and its synthesis probably takes place in the ER and the Golgi apparatus (Gardiner & Chrispeels, 1975).
If, as this work suggests, the bulk of the membrane protein in the ER and the Golgi bodies is not transferred, this would explain how these membranes and the plasmalemma maintain distinctive enzyme activities.
However, there is considerable evidence that the membranes in the Golgi body stack are moving rapidly through it. Certain unicellular algae, e.g. Prymnesium and Chrysochromulina, have only one Golgi body per cell. By way of compensation, there may be as many as 30 or more cisternae per stack. The organelle elaborates complex scales which coat the outer surface of the alga and the polarity of the stack is obvious from the gradation in complexity of scale precursors seen inside the cisternae in thin sections (Manton, 1966). This is the most direct evidence that whole cisternae are shuttled along the stack in one direction, from the ER, which apposes the immature ('proximal') end of the stack, to the plasmalemma with which the cisternae full of mature scales fuses. The Golgi body in higher plants and animals differs from that of these algae in that the cisternae vesiculate before fusing with the plasmalemma, but evidence from higher organisms also suggests the whole stack turns over. The number of cisternae per stack decreases in starvation or when 'secretory' vesicle production is stimulated and increases when vesicle production is inhibited (Morré et al., 1971). The alteration in staining properties across the Golgi stack affects the whole cisternal membrane and not just the peripheral vesiculations (Grove et al., 1968), and in isolated Golgi bodies this property spreads within a short time to the ER-like cisternae of the stack so that all the membrane resembles plasmalemma (Frantz et al., 1973).
Golgi-specific enzyme proteins could be retained in the organelle while the lipidic sack and plasmalemma-specific proteins pass on if there is movement of membrane protein between cisternae. The centre of the cisterna where it is closely appressed to its neighbours is the most likely site for this activity and the intercisternal space here is filled by a lipoprotein plaque (Mollenhauer et al., 1973), a hydrophobic environment in which lipophilic membrane proteins would be mobile. It is apparent from the staining evidence and the intermediary status of the Golgi body between ER and plasmalemma that there is a gradient in the concentration of substrate, protein and lipid, for the membrane-modifying glycosyltransferases of the Golgi, and the cisternal membrane at the proximal
pole of the stack has the highest concentration. Substrate binding alone could ensure that the net movement of Golgi enzymes is in the direction of the proximal pole. The glycosylation of the products, Golgi body-modified plasmalemma-specific proteins and lipids, would make them too hydrophilic to move rapidly between cisternae in this hypothetical scheme.
8.4.2—
Synthesis and Secretion of Extracellular Material
While investigations into the function of the Golgi apparatus in animals have concentrated on protein glycosylation and other membrane modifications, plant biochemists have been more concerned with a role for this organelle in polysaccharide biosynthesis.
Gardiner and Chrispeels (1975) have presented evidence that the glycosylation of the hydroxyproline-rich glycoprotein takes place in the Golgi apparatus. The completed polymer is a cell wall rather than a plasmalemma component and carries arabinosyl side-chains on its hydroxyproline residues. Organelle fractions were prepared on density gradients from carrot root tissue pulse-labelled with [14 C]-proline. Most of the label in hydroxyproline was associated with a Golgi body-enriched fraction. This fraction coincided with peak activity in the gradient of an enzyme which transferred arabinosyl residues from UDP-arabinose onto endogenous protein acceptors. (It should be noted, though, that the gradients used to prepare organelles contained glutaraldehyde and the distribution of enzyme activities on glutaraldehyde-free gradients is not strictly comparable).
From studies of thin sections, electron-microscopists proposed that the Golgi apparatus was involved in the synthesis and secretion of cell wall polysaccharides. The increase in luminal volume at the cisternal periphery was thought to represent the polymerization or decantation of a batch of wall material which travelled to the plasmalemma in a secretory vesicle and was discharged into the wall as the vesicle membrane fused with the plamalemma. This evidence has been critically reviewed by O'Brien (1972).
Much of the biochemical evidence has come from work on the root cap. It seems that to follow polysaccharide biosynthesis by autoradiography requires the extremely high rates of uptake and incorporation found only in certain differentiating or differentiated cells. The root cap is a very rapidly growing and metabolizing tissue—the entire cap of ca 10, 000 cells in maize is replaced every 24 hours. The cap cells secrete large quantities of slime to ease the passage of the cap through the soil. This material is a hydrated polysaccharide with a chemical composition similar to the matrix components of primary cell walls, pectin, but the structure of the polymer has been modified to reduce gelation, e.g. maize root slime-.polysaccharide has a substantial content of residues of fucose, a sugar absent from maize root pectin. Slime producing root cap cells each have several hundred Golgi bodies (root parenchyma cells have only ca 30) with a characteristic 'hypertrophied' morphology: the vesiculations of the cisternal periphery are very swollen.
Northcote and Pickett-Heaps (1966) fed [6-3 H]-glucose to wheat roots and analysed the pattern of incorporation of this label into polymeric material of root cap organelles by high resolution autoradiography. After 5 minutes of feeding, silver grains were confined mainly to the immediate vicinity of the Golgi bodies. By 10 minutes, both Golgi bodies and secretory vesicles were labelled, but very little radioactivity was associated with the wall. Subsequent incubation of labelled roots in non-radioactive glucose solution for 10, 30 and 60 minutes provided a time-series of sections showing progressive loss of radioactivity from the Golgi bodies and secretory vesicles and its accumulation outside the plasmalemma (Fig. 8.3). Chemical analysis of the high molecular weight polysaccharide in the root cap after 15 minutes exposure to radioactive glucose showed that more than 70% of the label in this material was in galactosyl residues. Galactose occurs only in pectin-type polymers in angiosperm cell walls and is the only major unit of these polysaccharides to retain a hydrogen atom on carbon 6. Since less than 3% of the polysaccharide label was in glucose units of a -cellulose at this time, the labelled material represents slime-polysaccharide or matrix components of the wall.
This is strong evidence that the Golgi apparatus is responsible for the secretion of pectin-type polymers. Though high molecular weight material appeared first of all in the Golgi apparatus, this does not establish that it was synthesized here. The early stages in polymerization must involve oligosaccharides which may be lost from the sections.
The differing functions of ER and Golgi apparatus in the synthesis and secretion of slime-polysaccharide in the root cap have been investigated by Bowles and Northcote (1972), and compared with the synthesis and secretion of the ordinary matrix components of the wall in the rest of the root. They fed [U-14 C]-glucose to maize roots and, taking different parts of the root, prepared organelle fractions by differential and 'step' density gradient centrifugation. These were characterized from thin sections. Radioactive polysaccharide was found in a rough ER fraction and a fraction rich in Golgi bodies. Chemically, the labelled material in membranes from the root tip resembled slime polysaccharide and that in membranes from mature root tissue had the composition of the cell wall matrix. It could have been slime or pectin from the wall which was mixed with and bound to, or enclosed in, membranes when the tissue was chopped. To test this, membrane fractions were prepared from unlabelled tissue chopped in an extraction medium with radioactively labelled, soluble slime and wall components. The membrane fractions were essentially unlabelled.
When [U-14 C]-glucose was fed to maize roots over times ranging from 5 to 90 minutes, radioactivity was incorporated steadily into slime, wall and membrane polysaccharide up to 30 minutes. After this time the slime and wall continued to accumulate label at the same rate but there was no further increase in the radioactivity of each of the sugar residues of the polysaccharide in membranes, confirming that these polymers are precursors of the slime and wall matrix (Bowles & Northcote, 1974).
Bowles and Northcote (1976) investigated this precursor material after labelling it to saturation from supplied [U-14 C]-glucose. Some of the polysaccharide was freely soluble with a high molecular weight (>40,000), most of the rest was so firmly membrane-bound it required protease digestion for release. These types were found in both membrane fractions, but the Golgi body-rich fraction had more of the former, whereas the ER had much more of the latter. Most of the membrane bound polysaccharide of the ER was as short chains (MW < 4,000), but all the polysaccharide segments bound to Golgi body membranes had molecular weight greater than 4,000. Interestingly, though all the other sugars of slime-polysaccharide were found in labelled residues of the short chains, fucose, which always occupies a terminal position on pectin side-chains, was absent.
The earliest polymeric precursors of polysaccharides will have the lowest molecular weights and it seems from this work that they are membrane-bound. The glycosyltransferases involved in chain-extension during pectin synthesis are also all bound to membranes (Villemez et al., 1965; McNab et al., 1968; Odzuck & Kauss, 1972). This early precursor is associated chiefly with the ER. Chain extension, addition of terminal fucosyl groups to slime polysaccharide and release from the membrane occur progressively as this material leaves the ER and passes through the Golgi apparatus and there is no evidence that synthesis is restricted to a particular section of the endomembrane complex.
ER fractions for both cap and mature root contained much more radioactive polysaccharide than the equivalent Golgi body fraction. However, since a whole-root ER fraction had 40 times as much lipid, the radioactivity in polysaccharide per unit quantity of membrane (i.e. weight of lipid) for the Golgi body-rich fraction was twice the value for ER. This could be because a smaller fraction of the ER is devoted to polysaccharide synthesis, or because the polymer chains are longer in the Golgi body, or both.
With the biosynthetic machinery saturated from [14 C]-glucose, Bowles and Northcote (1974) compared the rate of production of radioactive wall and slime with the steady-state levels in membrane polysaccharide. This enabled them to calculate the rates of turnover of polysaccharide in the membrane compartments expected for different models of secretion. For example, labelled wall polysaccharide is produced at 2,000 cpm per minute and the steady state level of radioactivity in wall polysaccharides in the Golgi bodies in 5,000 cpm. Therefore the entire polysaccharide content of the Golgi body will be replaced every 2.5 minutes if all the wall material has to pass through the Golgi apparatus.
Supposing polysaccharide and membrane sack move through the stack as a single entity, then for the average stack of 5 to 6 cisternae, one cisterna is released roughly every 0.5 minutes. This seems very fast, but it is remarkably close to values determined microscopically in other plants. Working with the Chrysophycean alga Pleurochrysis, Brown (1969) showed by time-lapse cinephotomicrography that the single Golgi body and all the other organelles rotate inside the cell wall, 360° in 15 to 20 minutes. Now the lateral displacement
observed in thin sections between successive cisternae released from the Golgi body was 15°, indicating that the time between the release of successive cisternae is about 0.75 minutes. Schnepf (1961) measured the volume of slime produced by glands on the leaves of the carnivorous plant Drosophyllum lusitanicum over set time periods. From the size and number of Golgi vesicles in the gland, he estimated that the rate of production of vesicles by each Golgi body necessary to maintain the observed rate of secretion was 3 per minute at 28°C.
Now there is good reason to suspect that 2 cisternae per min is an over-estimate of the speed required to shift all the matrix to the wall via the Golgi apparatus in maize roots. Firstly, while the recovery of slime and wall components is probably close to 100%, the recovery of Golgi bodies is certainly much lower and so the steady-state level of radioactivity in this organelle is underestimated. Secondly, the wall fraction from mature root tissue was shown to contain a large amount of labelled glucose polymer which was only a minor component in the membrane fractions. It could be cellulose or contaminant starch, but it means that the measured rate of increase in polysaccharide label of the wall fraction is probably an overestimate of the rate of production of wall matrix by the endomembranes. Nevertheless, observed rates of turnover of cisternal stacks can account even for this overestimate of the rate at which polysaccharide would have to pass through the Golgi body and there is no need, therefore, to invoke secretion via the vesiculating cisternal periphery independent of the turnover of the stack, or transfer direct from ER to the wall.
Similar calculations indicate that if slime-polysaccharide is secreted only via Golgi bodies, the entire polysaccharide content of these organelles is displaced every 20 seconds, a figure corrected for cellulose. The changing pattern of label in polysaccharide monomers after feeding [14 C]-glucose for different times confirmed that slime-polysaccharide was synthesized faster than wall matrix polymers. Some of it had been secreted from the cells within 2 minutes of supplying the labelled precursor. These results pay tribute to the furious synthetic activity of root cap tissue. Even using conservative estimates of the rate of turnover of cisternae, Morré et al. (1971) calculate that secretory vesicles contribute enough new membrane to the plasmalemma of a maize root cap cell to replace it entirely every 4 to 8 hours. How the excess is recycled is not known. Careful studies of Golgi bodies and numbers of secretory vesicles in thin sections of maize root caps fixed at different times have shown the existence of rhythmic fluctuations in secretory activity. The organelles reach a peak of activity every 3 hours, synchronized over the whole cap. Whole batches of roots can be synchronized by transfer to fresh solution, which induces a peak of activity at around 1 hour later (Morré et al., 1967). Since the results obtained by Bowles and Northcote refer to the first three quarters of an hour after transfer of roots to [14 C]-glucose solution, they probably represent peak activity for root cap Golgi bodies.
Though still in debate, it now seems unlikely that the cellulose microfibrils of the wall are synthesized in the ER or Golgi apparatus in higher plant cells
(see also chapters 1 and 7). Low incorporation into glucose relative to other wall monomers in the polysaccharides of ER and Golgi body-rich fractions of maize root confirms the earlier autoradiographic evidence from developing xylem vessels. In this tissue only the plasmalemma was labelled from [3 H]-glucose at the time it was being incorporated exclusively into cellulose (Wooding, 1968). However, Golgi body-enriched membrane fractions synthesized radioactive b -1,4 glucan when supplied with UDP-[14 C]-glucose (Van Der Woude et al., 1974; Shore & MacLachan 1975). The b -1,4 glucans synthesized from UDP-glucose have been shown to be cellulose and not b -1,4 glucan sections in glucomannan, a matrix component synthesized from GDP sugars (Villemez, 1974). Golgi body cellulose synthetase showed distinctive kinetics when compared with the activity in a plasmalemma-enriched fraction, and was much less active than the plasmalemma enzyme at high (1 mM ) substrate concentration. It seems, then, that the activity in Golgi bodies was not due to contamination by plasmalemma and that the Golgi apparatus may be ferrying the enzyme to the plasmalemma in a less active form. A similar mechanism has been shown for chitin synthetase in yeast and Mucor. Chitin is structurally similar to cellulose and is produced as a microfibril on the outer surface of the plasmalemma by an enzyme particle which spans this membrane. Chitin synthetase is made in an inactive zymogen form, found in microsomal membranes, which can be converted to an active enzyme by protease digestion. An inhibitor of protease found in the soluble cytoplasm is thought to control this transition, preventing the activation of the enzyme: before it reaches its operational site in the plasmalemma (McMurrough & Bartnicki-Garcia, 1973; Durán et al., 1975). The cellulose component of the scales of Chrysophycean algae appears to be made in Golgi cisternae, but this difference between them and higher plants may well amount to nothing more fundamental than the timing of cellulose synthetase activation.
8.4.3—
Differentiation
Inevitably the elements of the endomembrane complex are heavily involved in the differentiation of plant cells. Normal cell growth and development require the biosynthesis of intracellular membrane and extracellular wall material, both of which involve the endomembranes. When a cell differentiates the specialized nature of these products must be the result of altered biosynthetic activity of the endomembrane complex in response to information received, ultimately, from the nucleus. For example, by comparison with a parenchyma cell of the same root, slime-producing root cap cells synthesize more Golgi body membrane, more polysaccharide, and the biosynthetic machinery includes different enzymes, e.g. fucosyltransferases.
As the source of bulk membrane components the ER plays a unique role in the development of specialized organelles in differentiating cells. Undoubtedly the best characterized example is the development of glyoxysomes in castor bean. These organelles are found only in endosperm tissue and develop between
2 and 4 days after germination, by de novo synthesis of protein and lipid. Glyoxysomes do not contain the enzymes for phospholipid synthesis. When [14 C]-choline is supplied to endosperm tissue, the first membrane fraction to be labelled is the ER and this label subsequently appears as phosphatidyl choline in the glyoxysome membrane. In the early stages of germination, glyoxysomal enzymes can be detected in the ER fraction (Beevers, 1975). Because there is no evidence that glyoxysomes can synthesize protein, the kinetics of incorporation of radioactive amino acid into these organelles was interpreted as the result of synthesis in the ER followed by transfer to the glyoxysome. Thus the incorporation of [35 S]-methionine into glyoxysomes lagged 30 minutes behind the labelling of the ER. During a 'chase' with unlabelled methionine, the radioactivity in ER membranes decreased at the same time as the glyoxysome membrane continued to accumulate label (Bowden & Lord, 1976). In thin sections, the coincidence of a sheet of ER abutting end-on to a developing microbody is reported as frequent, both for glyoxysomes and for leaf peroxysomes. Although their membranes are seen to touch, the lumina of the two organelles appear quite separate. When segments of juvenile leaves are incubated in a solution of 3,3'-diaminobenzidine and H2 O2 (to localize catalase activity) and prepared for electron microscopy, the developing microbodies show a strong electron-opaque deposit but no such product can be detected in the contiguous ER (Gruber et al., 1973).
From electron-microscope sections, the vacuole of plant cells is thought to begin with the distension of portions of ER to form numerous 'pro-vacuolar' vesicles which then coalesce and further expand. A combined thin section and freeze-etch study of later stages in vacuole formation showed that each expanding pro-vacuole had a sheet of fenestrated ER lying close to its bounding membrane over the whole external surface (Fineran, 1973b).
These observations suggest that the microbodies and the vacuole develop in two phases. A primary event is the formation of a separate compartment from part of the ER. This pro-organelle must already differ from the bulk ER because its subsequent development is so different, but it is not known to what extent any of the components for its specialized function are present at this stage. Further development of this structure appears to be by transfer of lipid and protein from apparently undifferentiated ER close to or touching its bounding membrane.
Some correlations between the distribution of the ER and spatial patterns of wall biosynthesis in differentiating cells have suggested a morphogenetic role for this membrane. For example, in the differentiation of sieve-tubes, the siting of the sieve-pores in the end walls is pre-patterned by plates of ER, and in the differentiation of xylem vessels sheets of ER cover the internal surface of the plasmalemma between wall thickenings (Northcote, 1968). In both cases, the result of this pattern is a reduced deposition of secondary wall against the section of plasmalemma persistently overlain by ER. In many thin section electron micrographs of plant cells, large areas of plasmalemma appear to be covered closely by a continuous sheet of ER and it is difficult to see how
secretory vacuoles could get to the plasmalemma. Figure 8.3 shows secretory vacuoles apparently pouring through a gap in such a barrier. Juniper and Pask (1973) have shown that the deposition of slime polysaccharide is restricted to the outer tangential wall of maize root cap cells and their micrographs show that fenestration of the peripheral ER is most apparent on this side of the cell. The ER, therefore, may not only produce the polysaccharide of the wall but also control the site of its deposition. How the ER is organized in the correct pattern is a central problem of cell morphogenesis. Certainly the membrane is well placed to receive and is competent to interpret information from the nucleus and the possibility that it also 'responds' to chemical information from other organelles or outside the cell cannot be discounted.
The role of endomembrane complex in differentiation is undoubtedly not restricted to the 'motor' function of assembling new structures in response to a changing supply of nucleus-derived information. The effects of plant growth regulators in the tissue environment of the cell on nuclear activity must be mediated by the cytoplasm and, because of its position in the front line, the plasmalemma is strongly implicated. We know virtually nothing about the mechanisms involved in this 'sensory' function of the endomembrane complex. The first indication of the nature of this interaction may be the discovery that RNA polymerase in plants is activated by a factor released from plasmalemma by auxin (Cherry, 1974, see also chapter 13).
8.5—
Conclusions
The working definition of the endoplasmic reticulum is essentially negative. All the membrane of the cell not obviously specialized is relegated to this category. Biochemical investigation of its activities, stimulated and supervised by electron microscopy, has gone some way toward explaining the low level of its organization and replacing the negative definition with something more positive. The ER is the meristem of the cell. It is membrane in the process of synthesizing membranes, creating a pool of components and distributing them about the cell for the maintenance and enlargement of the specialized organelles. For this purpose the outer mem branes of plastids, mitochondria and the nucleus can be considered as rather more specialized 'limbs' of the ER, the two former membranes acting as placentas for the very specialized membranes they enclose. The new protein and lipid components must somehow cross the placental gap in a solubilized form. Material from the ER apparently contributes to the expansion of the tonoplast, microbodies and the plasmalemma in the same way (Fig. 8.6).
However, the glycoproteins, glycolipids and polysaccharides intended for the plasmalemma and wall are not transferred in this way. Probably they are too hydrophilic to be shuttled in and out of different sides of lipid bilayers. Once synthesized, they never leave the membrane (and its lumen) which is incorporated as a unit into the plasmalemma (and the wall). This vesicular transfer cannot
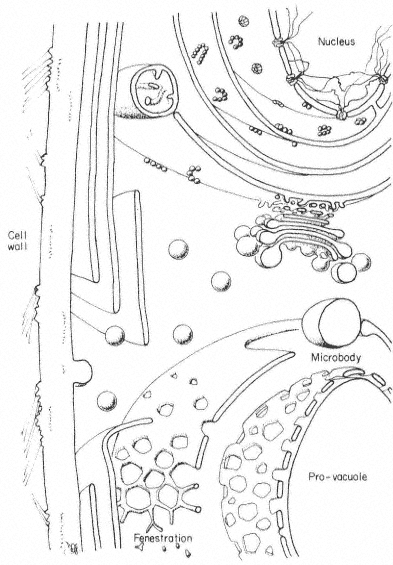
Figure 8.6
Diagram of a plant cell early in development showing the relationships
between the ER and the other membranes of the cell proposed
from electron microscope and biochemical evidence.
provide the selectivity of carrier proteins. Therefore all the membrane which is subsequently incorporated into the plasmalemma by fusion passes through a phase of close appression (the Golgi body) to allow the selective redistribution of membrane components.
In that the endomembrane complex is the cell's machinery for organelle and wall synthesis, the specialized nature of these ER products in differentiated cells can only come about as a result of reprogramming the synthetic machinery. Essentially it is the endomembrane complex which builds the plant cell and the nucleus which stores, selects and supplies designs for the appropriate tools.
Further Reading
Beevers H. (1975) Organelles from castor bean seedlings: biochemical roles in gluconeogenesis and phospholipid biosynthesis. In Recent Advances in Chemistry and Biochemistry of Plant Lipids, pp. 287–299 (eds. T. Galliard & E. I. Mercer). Academic Press, New York.
Bowden L. & Lord J.M. (1976) The cellular origin of glyoxysomal proteins in germinating castor bean endosperm. Biochem. J.154, 501–6.
Bowles D.J. & Northcote D.H. (1976) The size and distribution of polysaccharides during their synthesis within the membrane system of maize root cells. Planta (Berl. ) 128, 101–6.
Donaldson R.P. (1976) Merabrane lipid metabolism in germinating castor bean endosperm. Plant Physiol . 57, 510–15.
Morré D.J. & Mollenhauer H.H. (1974) The endomembrane concept: a functional integration of endoplasmic reticulum and Golgi apparatus. In Dynamic Aspects of Plant Ultrastructure, pp. 84–137 (ed. A.W. Robards). McGraw-Hill, Maidenhead.
Morré D.J. (1975) Membrane biogenesis. Ann. Rev. Plant Physiol.26, 441–81.
Northcote D.H. (1968) The organisation of the endoplasmic reticulum, the Golgi bodies and microtubules during cell division and subsequent growth. In Plant Cell Organelles, pp. 179–197 (ed. J.B. Pridham). Academic Press, London.
O'Brien T.P. (1972) The cytology of cell-wall formation in some eukaryotic cells. Bot. Rev. 38, 87–118.
Phillip E.I., Franke W.W., Keenan T.W., Stadler J. & Jarasch E.D. (1976) Characterization of nuclear membranes and endoplasmic reticulum isolated from plant tissue. J. Cell Biol.68, 11–29.