SECTION THREE—
THE MANIPULATION OF PLANT CELLS
Introduction
The ultimate objectives of any scientific investigation are twofold–to increase understanding, and to create opportunities for the application of the new knowledge, ostensibly in the service of man. The two objectives may not often be consciously connected in the minds of the investigators, but history and experience have demonstrated that new information always stimulates a drive towards practical application. This is already beginning to be true for plant cell biology through the directed manipulation of plant cells grown in culture.
As is described in chapter 15, it has been possible to culture organs, tissues and cells of higher plants under aseptic conditions for many years. These techniques, developed largely through assiduous application of the trial-and-error approach, are now becoming highly sophisticated and have already yielded information of great value to our understanding of cellular processes, particularly the cell cycle. In recent years, it has become possible to remove the walls from plant cells by enzymatic procedures, yielding naked protoplasts, some of which remain viable and may be propagated further (chapter 16).
The exploitation of cell and protoplast culture techniques for purely scientific purposes is still in its infancy, but already the methods have been used commercially for the propagation of valuable stock plants, and for the propagation of virus-free plants from virus-infected stocks. What makes cultured cells and protoplasts so exciting from the practical viewpoint, however, is the potential they appear to offer for artificial genetic manipulation. It is too early, yet, to say whether protoplast fusion or gene transfers (chapter 17) will yield new and revolutionary varieties of crop plants—the prospect does exist however, and a consideration of these matters seems a fitting note on which to conclude our coverage of the molecular biology of plant cells.
Chapter 15—
Plant Cell Culture
15.1—
Introduction
Almost every chapter of this book illustrates that research in plant cell physiology and biochemistry is, at each moment in time, limited by the resolving power of current experimental techniques and by the availability of appropriate experimental plant material. Here we are concerned with the development of new experimental materials (cultured organs, tissues and cells), with culture systems being evolved for their exploitation and with recent experimental work which illustrates that these culture systems are adding a new dimension to studies in plant cell physiology.
The complexity of higher plants has inevitably led workers, concerned with investigating particular aspects of plant physiology, to the use of systems which are of reduced complexity. Hence the early and continuing use by plant physiologists of isolated plant organs (e.g. seedling roots, leaves), complex tissue systems (e.g. discs cut from leaves and storage organs) organ segments (e.g. segments of coleoptiles and hypocotyls) and isolated cell organelles. However, isolated organs and organ fragments are still systems of very considerable complexity and they are, from the beginning, systems of declining viability and favourable sites for colonization by microorganisms.
The concept that the aseptic culture of isolated organs, tissues and cells would 'give some interesting insight into the properties and potentialities which the cell as an elementary organism possesses' and 'would provide information about the inter-relationships and complementary influences to which cells within the multicellular whole organism are exposed' was formulated as early as 1902 by Haberlandt. Then, after a lapse of more than 30 years, White (1934) described successful root cultures initiated from the root tips of tomato seedlings, and White (1939) and Gautheret (1939) demonstrated that the parenchymatous wound callus which frequently forms at the exposed surfaces of organ segments could be removed and grown indefinitely as a relatively undifferentiated tissue (callus) culture. From this period there has been rapid progress in organ and tissue culture. Root cultures have been developed from many species (Butcher & Street, 1964). Cultures of stem apices (meristem culture) and of leaf, flower and fruit primordia have been successfully established (Street, 1969). These organ cultures as experimental systems differ from isolated organs in two important respects; they are sterile (free from microorganism contamination) and are handled aseptically, and they are sytems where unimpaired viability is evidenced by growth involving both cell division and cell expansion. Such
cultures have contributed to our knowledge of the specific nutritional and hormonal requirements essential for the growth and development of the separate organs of the whole plant and of their specific physiology and biosynthetic activities (Street, 1969). Callus cultures have also now been established from a very wide range of species and have been used in studies on the initiation of root and shoot primordia (Street, 1975a), on cytodifferentiation (Wetmore & Rier, 1963, Gautheret, 1966; Torrey, 1971), on the induction of division in quiescent tissue cells (Yeomann & Aitchison, 1973), on the nature of plant tumour cells (Butcher, 1973) and on the synthesis of a diversity of secondary plant products (Yeomann & Aitchison, 1973).
In 1953, Muir reported that if fragments of callus cultures of Tagetes erecta or Nicotiana tabacum were transferred to liquid medium and agitated on a reciprocal shaker, then the callus fragments broke up to give a suspension of single cells and small aggregates of cells and that this suspension contained actively dividing cells and hence could be propagated by serial subculture (Muir, Hilderbrandt & Riker, 1954). Such liquid cell suspension cultures have now been obtained from calluses of a number of species and this chapter will outline the development of more sophisticated techniques for their culture and assess their value for studies on the control of growth, metabolism and differentiation in higher plant cells.
15.2—
Changes in Growth and Metabolism of Plant Cells in Batch Culture-Cytodifferentiation
Plant cells in batch culture, i.e. cultures in a fixed volume of culture medium, increase in biomass by cell division and cell growth until a factor in the culture environment becomes limiting and sends them into a stationary phase. When such stationary phase cells are subcultured they pass in succession through a lag phase, a short-lived period of exponential growth, a period of declining relative growth rate and then again enter stationary phase (Fig. 15.1A). Traditionally such cultures are initiated by an inoculum establishing a relatively high initial cell density and only accomplish a very limited number of divisions before entering stationary phase. For example cell cultures of sycamore (Acer pseudoplatanus ) initiated at ca. 2 × 105 cells ml–1 will reach a final cell density of ca. 3 × 106 cells ml–1 corresponding to 4 successive doublings of the initial population.
The degree of cellular aggregation in these cell cultures depends upon the species of cell, or cell line within a species, and the culture conditions, but always shows a basically similar pattern of change during the growth cycle of a batch culture. The culture at stationary phase contains the highest proportion of free cells and mean cell volume is at its maximum value. When subcultured to new
medium, the cells first embark upon a massive synthesis of new cytoplasm and associated organelles and then begin to divide. For a short time cell division proceeds at a specific growth rate (µ) which is constant and maximal (µmax ) for that culture environment. During this phase mean cell volume declines sharply and the proportion of cells in aggregates rises. Then the specific growth rate begins to decline (slowly at first and later at an ever increasing rate) mean cell volume increases and, associated with this cell expansion, the aggregates break up and release free cells. Associated with these growth and structural changes are changes in physiological activity. Measurements, on a per cell basis, of respiration, of the levels of individual cell constituents and of the activities of individual enzymes show that peaks of activity occur. These may be quite sharp and are not coincidental (Fig. 15.1A–D). Different metabolic patterns emerge and decline during the progress of batch culture. Thus RNA synthesis is initiated prior to cell division, proceeds for a time at a greater rate than cell number increase and then ceases whilst cell division is still proceeding (Short, Brown & Street, 1969; Nash & Davies, 1972). Free nucleotides (mainly UDP-glucose and ATP in sycamore cells) are synthesized rapidly during lag phase, presumably an essential preparation for subsequent synthesis of cell-wall polysaccharides and as an energy source for the endergonic processes of cell division, but their net synthesis ceases very shortly after the onset of division (Brown & Short, 1969). Similar transient high activity in carbohydrate oxidation by the pentose phosphate pathway during lag phase has been interpreted as providing the necessary NADPH for the massive biosynthesis achieved during the lag phase of the growth cycle (Fowler, 1971). By contrast, the very sharp peak in ethylene production in sycamore cell cultures occurs late in the cell division phase when the cells are beginning to increase in mean cell volume and may be responsible for initiating aggregate breakdown (Mackenzie & Street, 1970). An essentially similar pattern of ethylene production has been reported for cell cultures of Rosa spp., Glycine max, Triticum monococcum, Melilotus alba, Haplopappus gracilis and Ruta graveolens (La Rue & Gamborg, 1971). Large changes in the activity of phenylalanine ammonia-lyase (PAL) and in p -coumarate: CoA ligase occur prior to stationary phase in cultures of Glycine max (Hahlbrock, Kühlen & Lindl, 1971). A similar peak of PAL activity has been reported in cell cultures of Rosa sp. (Davies, 1972). These changes coincide with maximum production of total phenols by the cultures. Other secondary products are produced by cell cultures, the time of maximum synthesis being restricted to a phase in the growth cycle and often being markedly influenced in its intensity by the plant growth hormone composition of the culture medium (e.g. hemicelluloses and lignin by sycamore; see Carcellar, Davey, Fowler & Street, 1971; visnagin (a physiologically active furanochromone) by Ammi visnaga; see Kaul & Staba, 1967; caffeine by tea cell cultures; see Ogutuga & Northcote, 1970; various alkaloids, by cell cultures of solanaceous plants; see Tabata, Yamanito & Hiroaka, 1971). Thus during the progress of batch culture the cells pass through a series of contrasted physiological states which encompass cells
becoming meristematic, cells expressing high meristematic activity, cells undergoing expansion and becoming either metabolically quiescent or in which certain restricted metabolic pathways are emphasized.
Are these large changes in cellular structure and metabolic activity observed in batch-propagated cell cultures examples of cellular differentiation? This important question cannot at present be satisfactorily answered. Certainly cultured
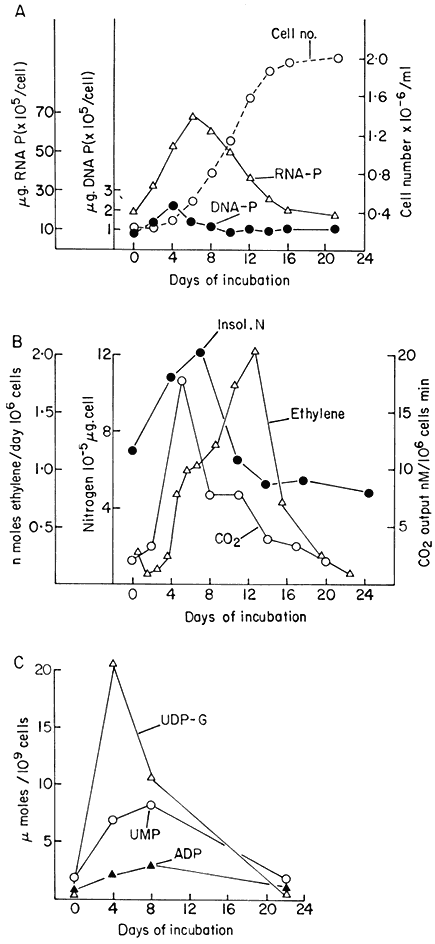
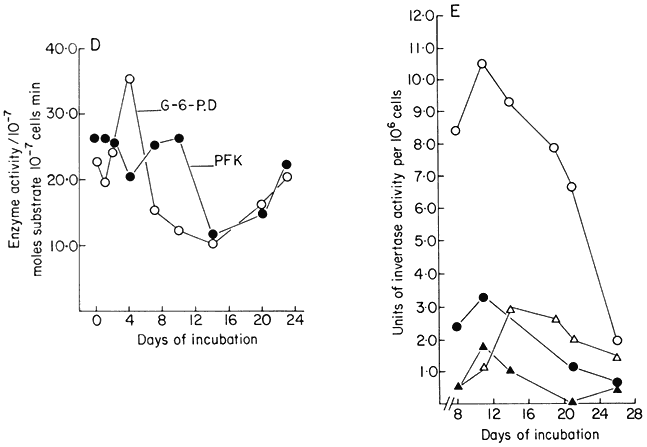
Figure 15.1
Changes in metabolic activity during the progress of growth of sycamore
(Acer pseudoplatanus L.) cells in batch culture. (a) Growth curve and data for
total DNA and RNA per cell (from Short, Brown & Street, 1969.) (b) Data for total
cellular nitrogen and respiration (from Givan & Collin, 1969) and for ethylene production
(from MacKenzie & Street, 1970.) (c) Data for levels of nucleotides (ADP=adenosine
diphosphate, UMP=uridine monophosphate, UDP-G= uridine diphosphate glucose)
(from Brown & Short, 1969.) (d) Data for activities of glucose-6-phosphate dehydrogenase
(G-6-PD) and phosphofuctokinase (PFK) (from Fowler, 1971). (e) Data for cell wall invertase
assayed at pH 4.5 (
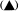
(from Copping & Street, 1972.)
cells do not correspond closely, either structurally or physiologically, with particular tissue cells of the plant body. To reproduce in culture the complete pattern of differentiation of selected specialized tissue cells is thus an objective not yet realized. Nevertheless the study of the origin in culture of particular physiological states and their cytological basis may advance our understanding of the molecular basis of cytodifferentiation in plants.
A recent book on cytodifferentiation in animal cells advances the widely accepted concept that the changes involved are consequent upon the activation of different sets of genes in different cell types and that this activation is expressed in terms of the synthesis of enzymes and other cellular proteins (Truman, 1974). Further, the differentiation process is regarded as a relatively permanent and irreversible change. In support of this it is possible to quote studies such as those of Cahn and Cahn (1966) on the culture of retina cells; such cells continue to produce the characteristic pigment granules during prolonged culture under conditions conducive to active cell division whereas in vivo such cells are non-dividing. If under certain secondary conditions of culture pigment granules were
lost, nevertheless when the cells were returned to the primary conditions of culture the pigmentation returned. To distinguish more minor and readily reversible changes in physiology from cytodifferentiation the former are described as modulations and are considered to reflect the operation of allosteric and other 'fine' processes of metabolic regulation. However, Truman (1974) concedes that 'it is not at all easy to draw a distinction between those enzymes which are fundamental to differentiation and those which represent very short-term modulations in the activity of cells' and in considering cytodifferentiation in liver cells states that 'differentiation and modulation do not represent distinct processes but are merely the extreme ends of a spectrum of changes that can occur'. Although from a number of plant species, cell cultures can be readily initiated from different organs (roots, stems, leaf petiole or lamina, cotyledons etc.) or from different living tissues within an organ (parenchymatous cells of pith, cortex, or mesophyll, cambial and other meristematic cells, immature vascular cells etc.) there is no very convincing evidence that they retain, in culture, characteristics of their in vivo origin although, as will be mentioned later in this chapter, they may not have undergone, during culture induction, the required degree of dedifferentiation necessary to express their totipotency (i.e. the capacity to generate a new plant in the way normally achieved from the fertilized egg). Such observations suggest that cytodifferentiation in higher plants, provided it has not proceeded to the point where cell death is inevitable, is a more readily reversible process than in the cells of higher animals. Hence the readily reversible physiological states observed in plant cell cultures may be basically identical with the states involved in normal cytodifferentiation. Further, it raises the possibility that cytodifferentiation does not depend on the transcriptional activity of different sets of genes for each kind of tissue cell but that the 'specialized' physiology of such cells may represent the influence of cytoplasmic factors (plant hormones?) on the stability and transport of RNA species and other aspects of the translational steps in gene expression. As discussed below, it may be possible to examine such hypotheses experimentally with plant cell cultures.
15.3—
Steady States of Growth and Metabolism of Plant Cells in Continuous Culture
Reference was made above to the short period of exponential growth observed in batch cultures. However, even during this phase of the growth cycle, the cells do not achieve a steady state (a state of balanced growth); cell division is uncoupled from increase in cell dry weight and protein content so that the cells are changing in size and composition despite the constancy of the double time of the culture (Fig. 15.2). Such observations raised the question of whether balanced growth could be achieved in plant cell cultures if a constant culture environment could be established by developing open continuous culture
systems (systems in which inflow of fresh medium is balanced by outflow of an equal volume of culture). Wilson, King & Street (1971) have developed such a system providing conditions of aeration and agitation appropriate to plant cell cultures, capable of long-term aseptic operation and functional either as a chemostat or as a turbidostat.
Figure 15.2
Unbalanced growth of sycamore cells (Acer pseudoplatanus L)
during the transient exponential growth phase achieved in batch culture.
(a) Semilogarithmic plots showing rate of change in cell number, total protein
and cell dry weight per unit volume of culture. The slopes of the lines of best
fit (calculated by linear regression analysis, P < 0.01) were used to determine
the specific growth rates (µ) for each parameter.
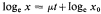
where x0 = initial value of parameter, x = value after time t(days). When
log10x plotted against t , slope = µ/2.303. (b) Changes in total protein
and cell dry weight per 106 cells with time calculated from data in A.
(From King & Street, 1973.)
15.3.1—
Chemostat Cultures
Subsequent work (King & Street, 1973; King Mansfield & Street, 1973) with this system operated as a chemostat (where equilibrium is established at a fixed rate of imput of a growth-limiting nutrient) has shown that long-term steady states of growth can be achieved with plant cell cultures and that such cultures conform to the chemostat theory developed from work with microorganisms (Monod, 1950; Novick & Sziland, 1950). In a chemostat, the relationship between cell density, x (cells per unit volume of culture), dilution rate, D (volume
of new medium added per unit time expressed as a fraction of the total culture volume), specific growth rate, µ (increase in biomass per unit biomass per unit time) and time (t ) is given by the equation
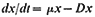
When equilibrium is reached and a steady state established dx/dt = 0, µ = D and x has a value characteristic of the dilution rate. Further, the nutritive environment remains constant, each nutrient achieving an equilibrium concentration which is related to its imput concentration and the rate of its consumption by the culture. As defined above, the equilibrium achieved in a chemostat culture results from one particular nutrient (depending upon the composition of the culture medium) becoming the limiting nutrient and determining the specific growth rate (µ) of the cells.
The nature of these steady states can be illustrated from work involving sycamore cell cultures growing in a synthetic medium (Stuart & Street, 1969) in which the supply of nitrogen is the limiting factor. Data for one such steady state (D = 0.194 day–1 ) is presented in Fig. 15.3. This shows that the cells are in a balanced state of growth and metabolism (as illustrated by the values for cell number, packed cell volume, cell dry weight, protein, DNA and RNA, and oxygen demand) and that the nutrient medium within the culture vessel is constant in composition (as illustrated by constancy of culture pH and the levels of glucose, phosphate and nitrate). Such steady state cells also display constant levels of metabolites (e.g. amino acids; Street, Gould & King, 1975) and constant levels of activity of individual enzymes (e.g. enzymes concerned with carbohydrate respiration; Fowler & Clifton, 1974; and with nitrogen assimilation; Young, 1973).
Such chemostat cultures can be operated from very low growth rates (i.e. low dilution rates) to growth rates approaching the maximum growth rate (µmax ) for the culture medium chosen, provided dilution rate is such that the cells can still achieve a matching growth rate. Of course if dilution rate is further increased, cell density does not stabilize and the culture suffers wash-out. Cells in balanced growth but highly contrasted in growth rate (and hence in cytology and metabolism) can therefore be obtained by chemostat culture. The range of change in certain cell parameters in sycamore cell cultures at different dilution rates (and hence specific growth rates) over the range D = 0.06–0.236 day–1 (corresponding to double times over the range 280–70 hr) is illustrated in Fig. 15.4. What this means is that it is possible to stabilize at will, by fixing dilution rate at an appropriate level, the individual physiological states which have only a transient existence in batch culture.
Work with chemostat cultures has shown that the same cell population can be taken through a series of steady states and then if the dilution rate is returned to that of an earlier steady state the cells again achieve not only the new predictable growth rate but also the physiological activities earlier recorded as
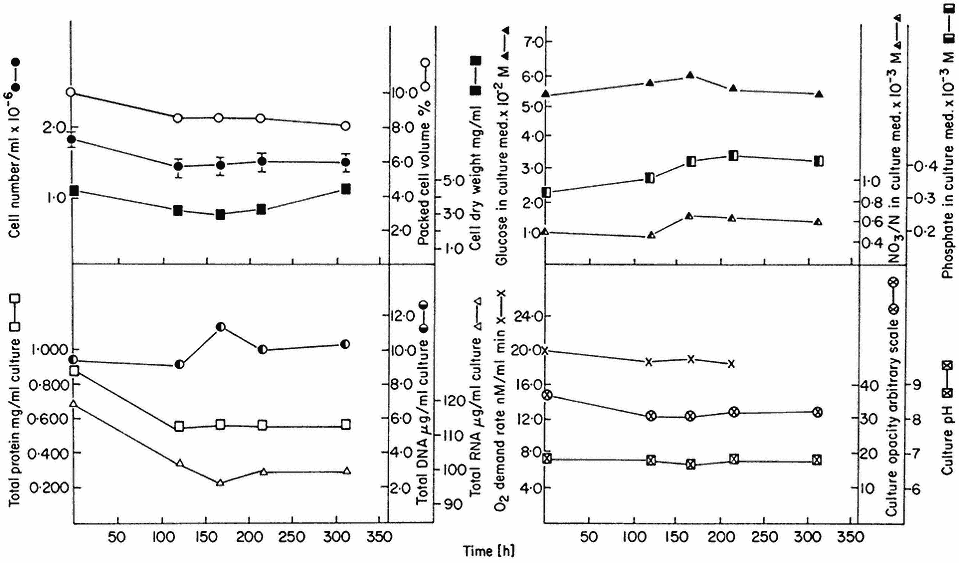
Figure 15.3
A steady state established in a 4-litre chemostat culture or A. pseudoplatanus cells. The
culture was diluted for 400 hrs. at a rate of 0.194 culture volumes per day. Samples were withdrawn
at intervals for biomass measurements, determinations of nutrient levels in the culture medium and for
respiration rate measurements. Culture opacity and pH were monitored continuously in the culture vessel.
(From King & Street, 1973.)
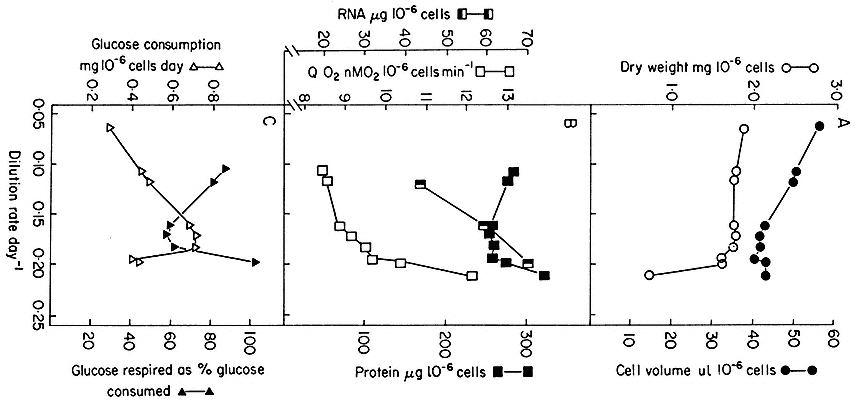
Figure 15.4
Steady state values for parameters of cell composition and physiological activities recorded
over a range of growth rates (as expressed by dilution rates) established in chemostat cultures
of sycamore cells (A. pseudoplatanus ) with nitrate/N as the limiting nutrient. The relationship
between dilution rate, D (fraction of culture volume displaced per unit time), specific growth rate
(m ) and doubling time (td) of the cell population in a steady state is given by
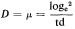
(From King & Street, 1973.)
characteristic. Such experiments demonstate the full reversibility of the cytological and physiological changes invoked. Interest will now focus on detailed studies of the kinetics of the transition between steady states in terms of enzyme activities and metabolite levels. Preliminary studies along these lines have already shown that during these transitions there are pronounced oscillations in enzyme activity levels (characteristic for each enzyme monitored) which gradually decline in amplitude as the new steady state is established. How far this behaviour is to be explained in terms of changes in rates of enzyme synthesis and degradation has yet to be determined. Clearly study of these transitions will yield entirely new data on metabolic regulation in higher plant cells; whether it will yield the key to expose the changes underlying cytodifferentiation is less certain.
15.3.2—
Turbidostat Cultures
The constancy of culture opacity in the steady state described by Fig. 15.3 has formed the basis for a second form of continuous culture—the turbidostat system (a continuous system in which inflow of new medium occurs in response to an increase in the opacity-decrease in the light transmission of the culture). The turbidostat culture system developed for work with sycamore cell cultures is shown in Fig. 15.5 (Wilson, King & Street, 1971). Here, each time the population density exceeds a pre-selected value as determined by the optical density continuously monitored by the photocell, an electronically operated valve opens to admit a pulse of new medium. This reduces the optical density of the culture and the valve closes. These imputs of new medium are balanced by periodic harvesting of small volumes of culture in response to an electronically controlled level detector which controls the output valve. The effect is to produce a culture of constant volume and constant population density growing at a constant rate. Whereas in the chemostat growth rate must always be below µmax (the culture growth being limited by the supply of a chosen nutrient) here in the turbidostat growth can safely proceed under non-limiting nutrient conditions and one can study the effect of physical factors (e.g. temperature, light regime, CO2 tension) and growth regulators, in particular plant growth hormones, on growth rate (King, 1976), and by appropriate techniques (Gould, Baylis & Street, 1974) on the duration of the different phases of the cell cycle. This is the system where one can attempt to achieve conditions under which biomass increase expresses the maximum genetic potential for cell growth. Such a system offers an entirely new approach to studies on the molecular basis of the hormonal control of cell growth.
15.4—
Synchronous Cell Cultures—Study of the Cell Cycle
As indicated earlier in this chapter, plant cell cultures are routinely propagated by batch cultures initiated at a relatively high initial cell density (2 × 105 cells
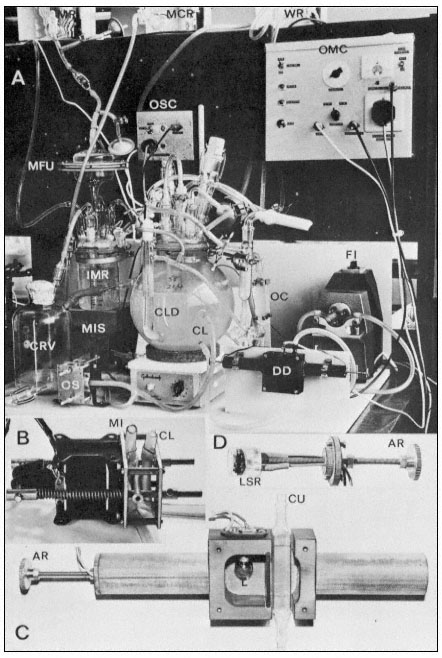
Figure 15.5
The turbidostat culture system of Wilson, King & Street (1971). The 4-litre culture vessel is mounted
over a magnetic stirrer and is controlled in temperature by an internal water circulating coil. The culture
flows through an external circulation loop (exit and entry lines of this loop labelled CL) by the action of
the flow inducer (FI). This circulation loop flows through a cuvette (Fig. C. CU) in the density detector
(DD). As the cells divide in the culture its opacity increases and this alters the light transmission between
the lamp (L) and the light sensitive resistor (LSR) in the density detector (see Fig. C and D). When this
transmission falls below a preset value the optical monitoring unit (OMC) sends an impulse to the medium
imput solenoid value (MIS) and a pulse of new medium flows from the intermediate medium reservoir (IMR)
fed from the main medium reservoir (MR) via a filter unit (MFU). The size of this imput of new medium is
controlled by the observation chamber (OC) in the circulating loop since the imput solenoid valve closes only
when new medium displaces culture in the cuvette within the density detector. These imputs of new medium
are balanced by release of culture into the culture receiving vessel (CRV) via the outlet solenoid valve (OS)
operated by an outlet solenoid control unit (OSC) responding to the electrodes in the constant level device
(CLD). Thus the culture is maintained at a predetermined optical density (corresponding to a fixed cell number
per unit volume) and the rate of entry of new medium (and balancing harvest of culture) is a measure of the
growth rate of the culture. Samples of culture can at any time be withdrawn for cell counting and biochemical
analysis via a sample tube located to the right of the culture vessel and after collecting the aliquot of culture
this sample collector can be washed with sterile water from the reservoir WR. Periodically the excess culture
collected in CRV is withdrawn and its volume measured and the outlet protected by washing with mercuric
chloride solution from the reservoir MCR. The various ports in the lid of the culture vessel provide for
introduction of the initial inoculum of cells, for aeration ( via a glass sinter tube) for withdrawal of samples
into the sample collector, and for flow of water through the internal temperature-controlling glass coil.
ml–1 ) and subcultured when they enter stationary phase. It is, however, possible to initiate such batch cultures of sycamore cells at much lower cell densities (the minimum inocular density being ca. 1.5–4.0 × 104 cells ml–1 in the standard medium; see Stuart & Street, 1969) and to use as inoculum cells maintained in stationary phase as long as possible without suffering decline in viability. When this is done using an enriched synthetic medium (Stuart & Street, 1971) the cultures show a more extended lag phase and then embark upon a succession of highly synchronous divisions as evidenced by the data for cell counts (Fig. 15.6), mitotic index (percentage cells in a recognizable stage of mitosis) determinations and estimations of nuclear DNA content (by microdensity following Feulgen staining) (Fig. 15.7; see also Street, King & Mansfield, 1971; Gould & Street, 1975). By using for such synchronous batch cultures the 4-litre culture vessel developed for the continuous culture systems but modified by adding a stainless-steel sampling valve automatically operated by a timing device (Wilson, King &
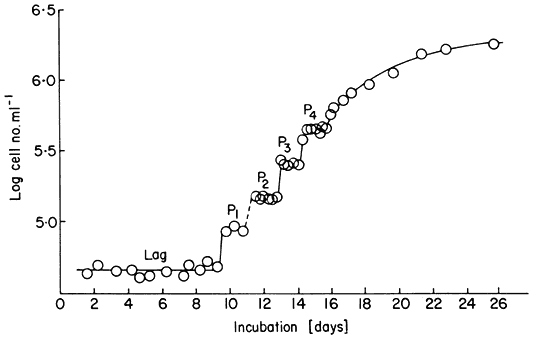
Figure 15.6
Growth curve of a synchronous cell culture of sycamore ( A. pseudoplatanus )
showing log cell number ml–1 with time. The interphase plateaux separating
the synchronous increases in cell number are labelled P1 –P4 . The lag phase of this
culture was of 9 days duration and the culture reached stationary phase on day 24.
Street, 1971) it has been possible to show the synchrony of a number of metabolic events in the cell cycle (King et al., 1974).
The period during which DNA is doubled takes place during a restricted period of the interphase between successive mitoses. Howard and Pelc (1953) termed this the S phase and distinguished the interphase period before S phase as G1 (the first gap) and the period after completion of S phase and before mitosis (M) as G2 (the second gap). Cell cleavage (cytokinesis) usually follows directly upon mitosis so that G1 can be timed from the origin of the daughter cells although, as originally defined, G1 begins as soon as nuclear division is complete. These stages in the cell cycle can be determined in exponential asynchronous cultures by determinations of doubling time (cell count data), mitotic index, labelling index (per cent nuclei and mitoses labelled following flash label with tritiated thymidine—3 H-Tdr), fraction of labelled mitoses (following pulse-labelling with 3 H-Tdr; see Quastler & Sherman, 1959; and continuous labelling with 3 H-Tdr; see Cleaver, 1967), microdensitomitry and autoradiography, achieved by the single slide technique of Mak (1965). These methods have been applied to cultures of sycamore cell lines (Gould, Bayliss & Street, 1974) during the phase of exponential growth in batch cultures and to steady state chemostat cultures growing at different rates (doubling times ranging from 22 to 85 hr). This has revealed that the phases S (7.0 ± 0.2 hr), G2 (8.7 ± 0.6 hr) and M (2.9 ± 0.3 hr) are relatively constant whereas, according to the cell doubling time, G1 varies widely (4–60 hr). This observation that G1 varies with different cycle times, whereas S + G2 is relatively constant has also been observed in work with mammalian cells both in culture and in vivo (Mitchison,
1971). Certain critical events in G1 may be essential to the initiation of S phase and may be rate limiting. In the work with sycamore cells growing at reduced rates, nitrogen supply is the limiting factor and when cells enter stationary phase in batch culture, they are arrested in G1 (this arrest is important in relation to the synchrony which can be achieved by regrowth of nitrogen-starved cells; see Gould & Street, 1975).
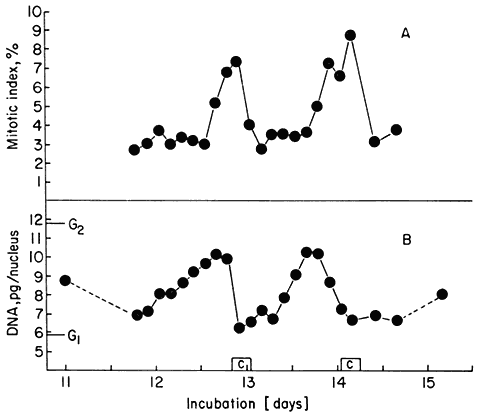
Figure15.7
(A) Mitotic index fluctuations for P2 and P3 of Fig. 15.6. (B) Average nuclear
DNA contents over the same period. G1 and G2 values for nuclear DNA
content of the cell line are shown for comparison. [C] represents the
durations of the two successive periods of cell number increase.
(Data from Gould & Street, 1975.)
The value of synchronous cultures for studies on the cell cycle is that they enable particular metabolic events to be monitored. Work along these lines with plant cells has only recently been undertaken. In a study involving synchronous sycamore cell cultures (King et al., 1974) it was shown that total extractable protein and RNA rise throughout interphase but at an increased rate during S + G2. This was supported by studies of the rate of incorporation of labelled amino acids and uridine into these fractions. Respiration rate similarly rose throughout interphase but with two peaks of activity during S phase and cytokinesis. When, however, changes in the activity of extracted enzymes were studied different patterns emerged. Thymidine kinase and aspartate transcarbamoylase showed single and separated peaks of activity (Fig. 15.8), succinic dehydrogenase showed two well separated peaks of activity, and glucose-6-phosphate dehydrogenase activity rose continuously throughout interphase.
Whilst the content of total extractable histone rose parallel with total protein, study of the rates of incorporation of 3 H-labelled lysine and 14 C-labelled arginine into this fraction pointed to greatly enhanced histone turn-over associated with S-phase (Street, Gould & King, 1975). It is not known whether the changes in enzyme activity detected in these studies reflected changes in rates of enzyme synthesis. Yeoman (1974) in work on cell synchrony in explants of Jerusalem artichoke tubers has shown however, by using the deuterium labelling technique (Hu, Bock & Halvorson, 1962), that changes in the activity of glucose-6-phosphate dehydrogenase during interphase do result from changes in rate of synthesis of the enzyme.
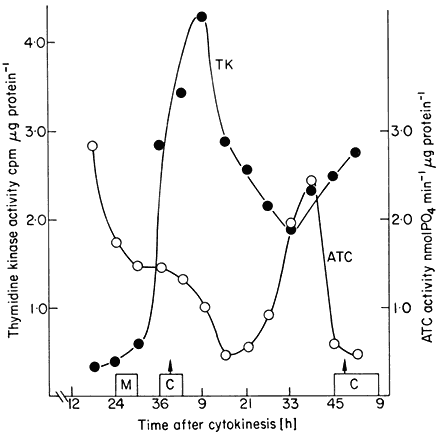
Figure 15.8
Changes in the activities of thymidine kinase (TK). and aspartate
transcamoylase (ATC) during a cell cycle in a synchronized cell
culture of sycamore (A. pseudoplatanus ). C = duration of cytokinesis
from the cell count data. M = duration of mitosis from the mitotic index data.
(From King, Cox, Fowler & Street, 1974.)
If future work along these lines enables the many separate metabolic events of the cell cycle of plant cells to be chartered, we will then have a number of 'markers' (points where particular metabolic events are initiated or terminated). This will enable us to identify those events along the interphase plateau whose initiation or pace is affected by nutritional factors and plant hormones, to determine whether certain events only occur when their controlling genes (which could be 'mapped') are exposed during DNA replication and whether other processes which proceed continuously show gene dosage effects as
evidenced by increases in their rate after the points in DNA replication when their controlling genes are duplicated.
15.5—
Morphogenesis in Cell Cultures—Concepts of Totipotency and Determination
Various forms of morphogenesis have been observed in cell suspension cultures; this morphogenesis is apparently dependent upon the development in the cultures of relative large aggregates of cells, often several hundred in number, associated in a symplast. The morphogenesis can take the form of root initiation, shoot bud initiation or the development of somatic embryos (often referred to as embryoids) (Konar, Thomas & Street, 1972). Usually only one form of morphogenesis is expressed; sometimes the situation is more complex but in these cases one form is usually dominant—carrot cell cultures can be manipulated to show predominantly root initiation or exclusively embryoid development (Kessel & Carr, 1972).
15.5.1—
Somatic Embryogenesis
Work on embryogenesis in carrot cultures illustrates clearly the problems in cell physiology raised by the morphogenetic potential of plant cell cultures. These studies date from the report in 1958 (Steward, Mapes & Mears, 1958; Steward, 1958) of embryo-like plantlets in liquid carrot cultures and their continuing development when the cultures were plated out on agar-solidified medium. Further work quickly established the presence in such cultures of structures strikingly similar to the globular, heart-shaped and torpedo-shaped stages of normal embryology from the zygote.
Halperin and coworkers (Halperin & Wetherell, 1964; Halperin, 1967; Halperin & Jensen, 1967) and Street and coworkers (Smith & Street, 1974; McWilliam, Smith & Street, 1974) have shown that the embryos in carrot cultures arise from single cells at the surface of the cellular aggregates and are released, at various stages of development, as free-floating structures which, if they have already reached the advanced globular stage, are capable of completing their development in isolation from the parent embryogenic clump. These cultures grow actively, the embryogenic clumps proliferating and fragmenting due to enlargement and separation of cells in their interior, and do not form embryos in a synthetic medium containing sucrose, inorganic salts, thiamine, meso -inositol, kinetin and auxin (2, 4-dichlorophenoxyacetic acid). To initiate embryogenesis they are transferred to a similar medium, lacking auxin, which contains nitrate and ammonia (or urea or glutamine) and is at pH 5.0–5.4. Despite earlier claims, coconut milk is neither essential for growth nor for embryogenesis. Evidence that immature embryos have exacting nutritional
requirements (isolated embryo culture—see Street, 1969) and the contrasted simplicity of the culture medium which supports prolific embryogenesis in carrot cultures suggests that the embryogenic cell aggregate fulfils a 'nurse' role to the embryogenic surface cells and that the embryos must remain attached to the aggregate to complete their early development to the point where they can survive and mature into plantlets when released.
This phenomenon of somatic embryogenesis raises a number of questions of cellular physiology. The term totipotency has been introduced to describe the embryogenic competence of the single cells from which the embryos arise. At present it is only possible to establish new plants, whether via embryogenesis of via shoot bud initiation, followed by adventitious root development, from the cell cultures of a limited (if now quite large) number of species or varieties within species. Cultures which show this morphogenetic potential support the view that the pathways of cytodifferentiation which result in living tissue cells do not involve any loss or permanent inactivation of the genome and that they are, under appropriate environmental stimuli, completely reversible. Whilst however some cell cultures remain recalcitrant, this cannot be established as a universal principle. It may be that in such cases the conditions of culture fail to provide or permit the synthesis of an essential morphogen. Halperin (1967) has, on the other hand, advanced the very interesting hypothesis that the achievement of totipotency occurs during the initiation of the carrot culture from the primary explant (storage root or seedling organ) and that embryogenesis is expressed by cell clumps derived from such 'induced' cells, the primary culture consisting of both these and 'non-induced' cells. Retention of high embryogenic capacity in the cultures will then depend upon culture conditions favouring the active proliferation of the induced cells. On this hypothesis, failure to obtain embryogenesis in culture would have as its primary cause inappropriate conditions of callus initiation; the conditions of initiation would have effectively activated cell division and growth in the explant cells but failed to achieve the necessary 'dedifferentiation' to obtain cells with the competence of the zygote (a concept already raised here in previously discussing cytodifferentiation in cell cultures).
In classical plant embryology it has been considered that the early segmentations of the zygote (at least up to the 16-celled proembryo) follow a precise and species specific sequence which has phylogenetic significance (Johansen, 1950). During these divisions the cells of the proembryo are considered to inherit different cytoplasmic potentialities from the different regions of the zygote and these differences are regarded as determining from the beginning the exact role they and their daughter cells will play in constructing the embryo and its parts. This concept is often referred to as the theory of precise mosaic organization. The early segmentations involved in somatic embryogenesis have now been followed in a limited number of species (Street, 1976). Figure 15.9 illustrates the sequence of early embryology in carrot cultures. These segmentation sequences involved in somatic embryogenesis show more uniformity one with another than is depicted in the published accounts of the zygote embryology
of the species concerned. In summary, early development of somatic embryos involves enlargement and regular segmentation in a subspherical cell mass by walls of minimum surface. This supports the concept of D'Arcy Thompson (1942), based upon his studies in animal embryology, that surface tensions are important in determining the early segmentations of embryology and that only at a later stage do localized growth centres emerge whose functioning gives rise to the divergences in the morphology and anatomy of embryos of different species. This regulative theory of organization regards the early segmentations as being controlled by physical factors and as not involving any 'determination' of the early formed cells. With this background, the greater diversity and specificity recorded for zygotic embryogenesis can be interpreted as resulting from the physical restrictions and polar chemical gradients imposed upon the embryo as it develops within the ovule; when these influences are removed, as in cell cultures, the embryology reverts to a more basic or 'primitive' type of segmentation. This interpretation is supported by observations on the segmentations observed in natural polyembryony and by recent reports that indeed much more variable patterns of segmentation occur in ovule embryology than has hitherto been recognized (Jensen, 1965; Brown & Morgensen, 1972).
15.5.2—
Polarity of Embryogenic Cells
The recognition that the embryos arising in cell culture have their origin in superficial cells of the cell aggregates raises the question of whether such cells have any unique cytological characters and whether their observation can yield information on the physical basis of cell polarity. In carrot cultures proliferating in the presence of 2, 4-D (Street & Withers, 1974) these cells are small, rich in cytoplasm and with a large diffusely-staining nucleus containing a prominent nucleolus. Small vacuoles are clustered round the nucleus and each cell contains several amyloplasts containing prominent starch grains (Fig. 15.10). Study of these cells in the electrorn microscope shows the presence of numerous round and oval mitochondria and Golgi bodies and the regular presence of small numbers of lipid bodies (spherosomes). As might be expected from their meristematic activity, these cells are frequently observed in mitosis (in contrast to the expanded interior cells of the aggregates) and show numerous wall microtubules and limited arrays of cytoplasmic and nuclear microfibrils. When the cultures are transferred to auxin-free medium there is a transient increase in proliferation prior to the initiation of embryogenesis and the superficial cells show a change in segmentation pattern leading to the origin of 4-celled groups (Fig. 15.10). The individual cells in these groups either initiate an embryo, or by their further division promote the growth of the aggregate or undergo expansion (and senescence?) and become involved either in the release of the developing embryo or the break-up of the proliferating embryogenic aggregate. Associated with this changed segmentation pattern the densely cytoplasmic superficial cells
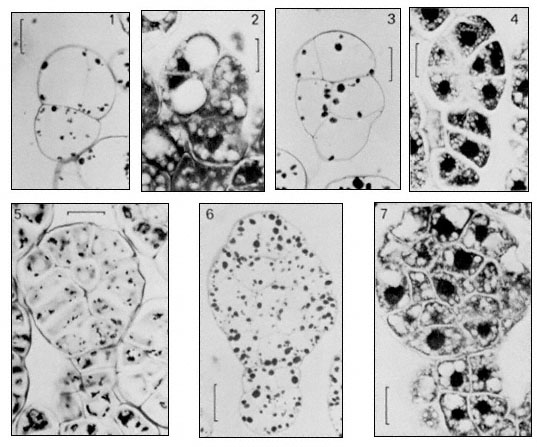
Figure 15.9
Earlier stage in the development of somatic embryos as observed in a cell suspension
culture of carrot (Daucus carota, L ). Scale lines on 1–4 = 20 m m, on 5–7 = 25 m m. Stages 1, 3,
5, 6 stained with periodic acid—Schiff (PAS), stage 2, 4 and 7 stained with toluidine blue (TB).
(From Street & Withers, 1974.)
show changes in fine structure. They have increased numbers of E.R. profiles (parallel arrays of rough E.R. profiles become particularly prominent) and of ribosomes. The Golgi bodies are more numerous and more compact. Additional large mitochondria (discs with swollen rims in outline) make their appearance and may come to occupy a considerable volume of the cytoplasm. The cells of the young proembryos show similar fine structure (Fig. 15.11). Although the first division of the embryogenic cells is by a wall parallel to the surface of the aggregate and at right angles to the longer axis of the cell (giving rise to an apical and a basal cell; the former being the first cell of the proembryo proper and the latter of the very variable suspensor), nevertheless fine structure studies do not reveal any prior asymmetry in the distribution of cytoplasm or cell organelles. Rather disappointingly, these studies have not revealed any unique features of the embryogenic cells or exposed any structural polarity. Perhaps this is not unexpected when we bear in mind the lack of any uniformity of fine structure in those angiosperm zygotes which have been studied (Jensen, 1965; Schulze & Jensen, 1969; van Went, 1970; Morgensen, 1972). Such studies only serve to emphasize that the special nature of embryogenic cells must now be approached at the level of molecular biology.
15.6—
Pollen Grains as Isolated Embryogenic Cells and as a Source of Haploid Cell Lines for Mutagenesis
This field of study was opened up by the pioneering studies on anther culture by Guha and Maheshwari (1964) working with Datura, and Bourghin & Nitsch (1967) working with Nicotiana. They showed that anthers, excised at an appropriate stage in pollen grain development and cultured in a simple medium gave rise to haploid embryos derived from individual pollen grains. Haploid plantlets can now be obtained by this technique from many species within the family Solanaceae. This approach extended to species in other angiosperm families has in a few cases yielded haploid callus but more frequently given a negative result. If we assume that immature pollen grains can, under appropriate stimuli, embark upon embryogenesis (sporophyte development) then the difficulty of achieving this with most excised anthers may be because conditions within the anther are too strongly promotive of the gametophyte pathway of development (i.e. that which gives functional pollen grains containing a tube nucleus and a generative cell). Recently however Nitsch (1974) in work with two Solanaceous species Nicotiana tabacum and N. sylvestris has shown that if anthers are excised at the peak of the first pollen mitosis, kept at 5°C for 48 hours and cultured for 4 days in a simple medium, then the immature pollen grains can be extracted. If this pollen grain suspension is cultured in a thin layer of liquid medium containing IAA, zeatin, glutamine and serine as supplements, some 10% of the grains embark upon embryogenesis. Although this work has so far
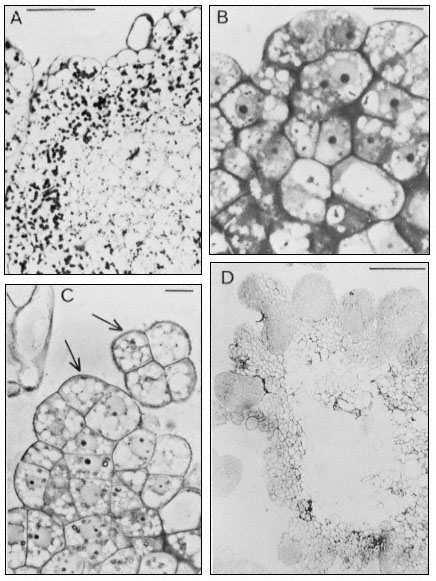
Figure 15.10
Embryogenic cell aggregates in suspension cultures of carrot ( D. carota ).
A and B from cultures in medium containing 0.1 mg 1–1 2,4-dichlorophenoxyacetic
acid (2,4-D), C and D from cultures in media with 2,4-D omitted. (A) Section through a
cell clump stained with PAS. Scale line = 200 µm. (B) Section stained with TB to show the
densely cytoplasmic superficial cells with nuclei surrounded with small vacuoles and more
internal cells with a large central vacuole. Scale line = 25 µm. (C) Localized superficial region
of active cell division. Arrows indicate characteristic pattern of cell segmentation which appears
following subculture to the auxin-free medium. Scale line = 10 µm. (D) General view of section through
a cell aggregate bearing numerous globular embryos at its surface. Stained with TB. Scale line = 200 µm.
(A & B from Smith & Street, 1974; C & D from Street & Withers, 1974.) (See also legend to Fig. 15.9.)
been restricted to Solanaceous plants, it could prove to be the key to inducing embryogenesis or haploid callus initiation in pollen grains of the many species resistant to the culture of whole anthers.
The importance of these studies is twofold. First is their potential for providing haploid plants (and by appropriate treatment homozygous diploids) and haploid cell cultures from many species and varieties. The haploid cell lines could be used as a source of mutant cell lines extending dramatically the field of the biochemical genetics of higher plants. Such mutant haploid lines could be rendered diploid by colchicine treatment and hence used to generate fertile mutants, provided their potential for morphogenesis was not impaired by the mutation. They could be preserved as cell culture lines by freezing preservation (Nag & Street, 1973, 1975a,b) as is already routinely done with mutant lines of bacteria. The use of such mutant cell lines in the continuous and synchronous culture systems previously described would permit more critical studies to be undertaken on particular metabolic sequences and their control and on the biochemical nature of the controls which operate in cell growth and division. Work along these lines is already actively proceeding (Widholm, 1974; Street, 1975b) but is currently handicapped by the need for more effective means of selecting the desired mutant cells which are present in very small numbers in the cell populations after mutagen treatment (e.g. 1 in 107 ).
The second important feature of these studies is in relation to embryogenesis. If within the pollen suspension, the pollen grains destined to embark upon embryogenesis are of characteristic size and/or density, it may be possible to isolate them by appropriate density gradient centrifugation. This would enable us to characterize embryogenic cells by the techniques of molecular biology, and to study in greater detail the segmentation patterns of early ermbryogenesis and the spontaneous expression of polarity in the proembryonal cell mass. Since the early stages of embryogenesis in pollen occurs within the enclosing pollen grain wall, study of the food reserves of such grains and of their biosynthetic activity may enable us to determine the special requirements of the proembryo.
This chapter has drawn attention to the availability of new experimental material and of new techniques in the field of plant cell culture. The exploitation of this approach in the molecular physiology of plants is still in its infancy. It is therefore important that the new generation of plant physiologists and biochemists should remain well informed of its present transient limitations and assured future prospects.
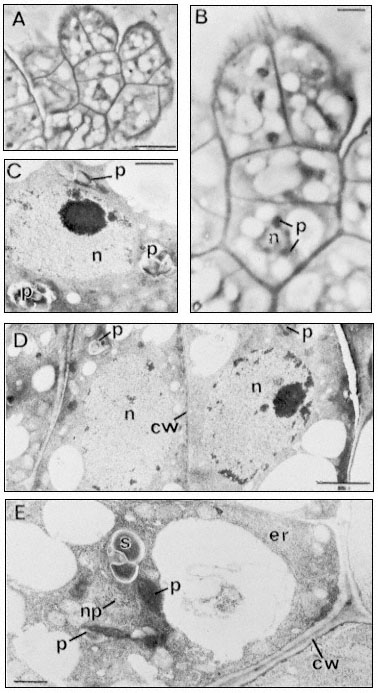
Figure 15.11
Light (A and B) and electron micrographs (C,D,E) of a four-celled somatic
embryo of D. carota. (A) The embryo located within a characteristic small
cell group (compare Fig. 10, C) at the surface of an embryogenic cell aggregate.
Scale line = 10 µm. (B) Enlarged view of embryo from A. The nucleus (n) of the basal
cell is seen to be surrounded with optically dense plastids (p). Scale line = 2.5 µm. (C)
The nucleus of the middle cell of the embryo surrounded by amyloplasts (p). Scale line
= 1 µm. (D) The two terminal (apical) cells of the embryo showing large rounded nuclei,
amyloplasts and the newly-formed thin dividing wall between them. Scale line = 2 µm. E.
The basal cell of the embryo separated from adjacent cells of the aggregate by a thick
plasmodesmata -free wall (cw). Parallel arrays of endoplasmic reticulum (er) can be seen
at the cell periphery. The nucleus (note glancing section showing nuclear pores–np)
is again surrounded by amyloplasts (p). Scale line = 1 µm. (From Street & Withers, 1974.)
Further Reading
King P.J., Mansfield K.J. & Street H.E. (1973) Control of growth and cell division in plant cell suspension cultures. Canad. J. Bot.51, 1807–23.
Ledoux L. (Ed.) (1975) Genetic Manipulations with Plant Material. Plenum Press, London. Les Cultures de Tissus de Plantes. (1971) Colloques Internationaux du C.N.R.S. No. 193, Paris.
Steward F.C. (Ed.) (1969) Plant Physiology. Vol. 5B. Academic Press, New York.
Street H.E. (Ed.) (1977) Plant Tissue and Cell Culture. Blackwell Scientific Publications, Oxford. (Second Edition.)
Street H.E. (Ed.) 1974) Tissue Culture and Plant Science, 1974. Academic Press, London.
Street H.E. (1976) Experimental embryogenesis—the totipotency of cultured plant cells. In The Developmental Biology of Plants and Animals. (eds Wareing, P.F. and Graham, C.F.). Blackwell Scientific Publications, Oxford.
Wilson S.B., King P.J. & Street, H.E. (1971) Studies on the growth in culture of plant cells. XII. A versatile system for the large scale batch and continuous cultures of plant cell suspensions. J. exp. Bot.21, 177–207.
Chapter 16—
The Physiology of Isolated Plant Protoplasts
16.1—
Introduction
The isolated plant protoplast is a single cell, bounded by the plasmalemma and, when first formed, containing all the normal cell components, although being separated completely from its cell wall. From a physiological viewpoint, however, the protoplast cannot be regarded simply as a cell lacking a wall, since the mechanics of isolation, in conjunction with environmental factors, undoubtedly influence its metabolism and elicit subtle ultrastructural changes. The absence of a functional cell wall may affect the permeability of the cell membrane and lead to a general leakage of solutes from the protoplast. The protoplast is also in a transient state since most protoplasts, irrespective of their immediate cultural environment, will initiate the synthesis of a new cell wall a few hours after release, and eventually revert to a single-walled cell. In spite of these considerations, and combined with a cautious extrapolation of experimental data to the presumed situation existing in the intact cell or whole plant, protoplasts provide an important biochemical tool for the biologist.
In the absence of a cell wall, the exposed plasmalemma can be examined in great detail with respect to particle uptake, permeability, possible membrane-associated functions such as disease resistance, membrane fusion, and, during cell wall synthesis, the relationship of the plasmalemma to its cell wall.
16.2—
Isolation and Culture of Protoplasts
16.2.1—
Preparation of Protoplasts
Protoplasts may be produced, under aseptic conditions, from a wide range of plant species either directly from the whole plant, or indirectly from in vitro cultured tissues. There are two basic approaches for the enzymatic isolation of protoplasts: (a) the treatment of a plant tissue with a mixture of pectinase and cellulase so as simultaneously to macerate, or separate, cells and degrade their walls (Power & Cocking, 1970); and (b) the sequential (two-step) method involving the production of isolated cells which in turn are converted into protoplasts by a cellulase treatment (Nagata & Takebe, 1970).
Since removal of the cell wall results in loss of wall pressure upon the cell, protoplasts are isolated and maintained in hypertonic plasmolytica provided by a balanced inorganic salt medium or monosaccharide sugar solution. Mannitol,
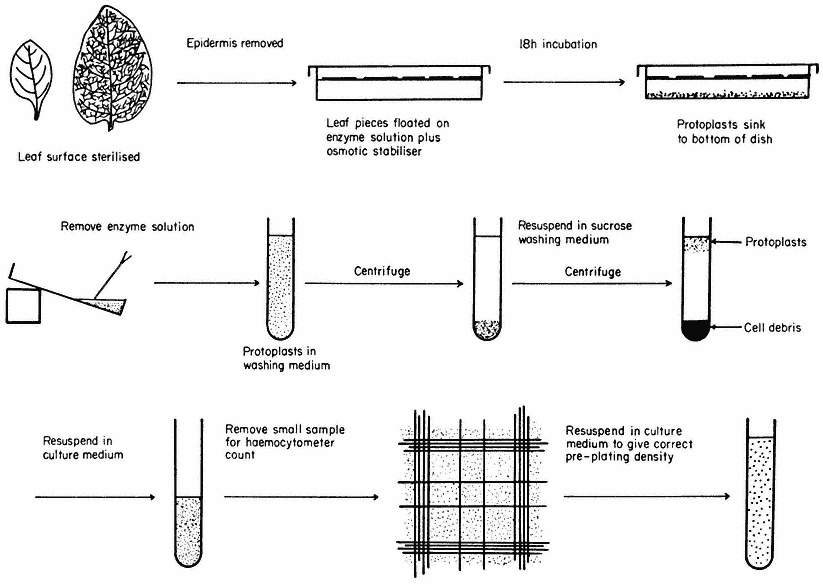
Figure 16.1
Generally applicable scheme for the isolation, washing and counting of leaf mesophyll
protoplasts. Protoplasts isolated from callus or cell suspensions are handled in the same way.
for example, is not readily transported across the plasmalemma and therefore provides a stable osmotic environment for the protoplast.
The enzymes used for the isolation of protoplasts are crude extracts of fungal origin. The pectinases are rich in polygalacturonidase activity whilst the cellulases contain hemicellulase, b -1,4-glucanase, chitinase, lipase, nucleases and pectinase.
A generally applicable scheme for protoplast production, using the mixed enzyme procedure, is shown in Fig. 16.1. In order for the enzymes to gain access to the plant tissues, as in the case of leaf palisade and mesophyll cells, the lower epidermis must be removed by peeling or partial digestion with cutinase. Certain types of leaves, particularly of the cereals, must be sliced prior to enzyme incubation, since the epidermis cannot readily be removed. Most plant tissues and organs (roots, root nodules, coleoptile, leaf epidermis, petals, germinating pollen grains, fruit placenta, tetrads and microsporocytes) will yield protoplasts after suitable adjustment of the enzyme mixture and plasmolyticum. For example, the pollen tetrad wall consists of callose and so only an enzyme rich in b -1, 3-glucanase (snail digestive juice enzyme) will liberate protoplasts.
Protoplasts are produced from calluses and cell suspensions (Fig. 16.2b) (Wallin & Eriksson, 1973) often only during the log phase in their growth cycles, since the composition of the primary cell wall varies as secondary products, such as lignin, are deposited as the culture matures, rendering it unsusceptible to complete degradation by cellulase. In general, the production of protoplasts from an untried source will always involve a consideration of enzyme purity, pectinase to cellulase ratios, protoplast yield and viability.
Following enzyme incubation, the spherical protoplasts (Fig. 16.2a, 16.2d) must be separated from cellular debris, subprotoplasts and vacuoles. Subprotoplasts are formed during early plasmolysis when the protoplast splits into two or more subunits, some of which will be enucleate and hence non-viable. Separation can readily be achieved in a variety of ways: repeated resuspension and centrifugation of protoplasts in a washing medium; flotation of protoplasts on a hypertonic sucrose solution (Fig. 16.1); passage of the incubation mixture through a nylon sieve of suitable pore size which allows only protoplasts to pass through; or by the use of a two-phase system, such as dextran-PEG (polyethylene
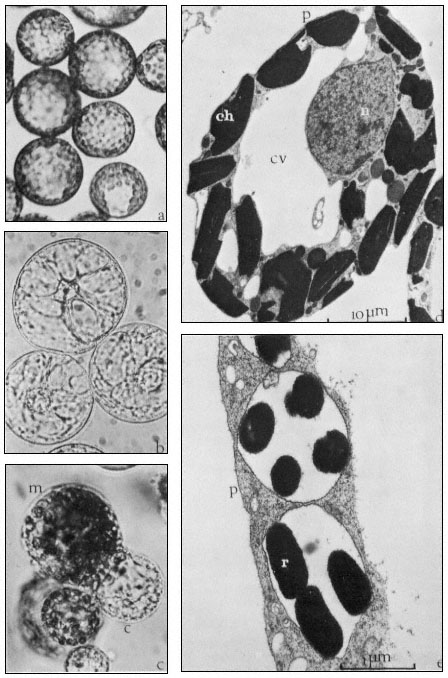
Figure 16.2
(a) Freshly isolated mesophyll protoplasts (40 µm diam.) in liquid medium
(Petunia hybrida). (b) Protoplasts (60 µm diam.) released from cultured cells. The
centrally positioned nucleus is surrounded by cytoplasmic strands ( Parthenocissus
tricuspidata). (c) Sodium nitrate induced fusion between a mesophyll (m) protoplast,
containing chloroplasts, and a colourless (c) protoplast isolated from a cell suspension.
Plastids are seen entering the cytoplasm of the colourless protoplast. (d) Electron micrograph
of a freshly isolated mesophyll protoplast (n = nucleus; ch = chloroplast; cv = central vacuole;
p = plasmalemma). (e) Uptake of Rhizobium (r) into vesicle within the cytoplasm of a protoplast
(p = plasmalemma). (Electron micrographs provided by Dr. M.R. Davey.)
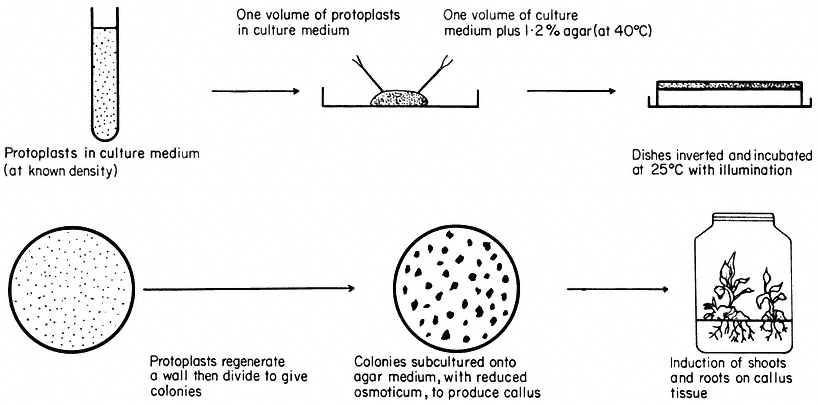
Figure 16.3
Plating of protoplasts (whole plant or cultured cell origin) in agar solidified
nutrient medium. Whole plant regeneration from protoplasts takes approximately five months.
glycol), which is based upon a density difference between protoplasts and cells (Kanai & Edwards, 1973).
Freeze etched protoplasts, collected after washing and examined in the electron microscope or treated with fluorescent brighteners which specifically bind to cellulose, reveal the complete absence of cellulose fibres on the plasmalemma surface.
16.2.2—
Cell Wall and Whole Plant Regeneration
Protoplasts are cultured in either liquid or agar solidified nutrient media (Fig. 16.3). The culture media are very similar in composition to those required for the in vitro culture of cells, but with the addition of an osmotic stabilizer to prevent bursting. During culture, up to 90% of protoplasts isolated from highly differentiated cells undergo a rapid and immediate process of dedifferentiation whilst at the same time initiating the synthesis of a new cell wall.
Early stages of wall synthesis are preceded by extensive infoldings of the plasmalemma together with an accumulation of pectin-like substances in vesicles found in the peripheral layer of cytoplasm. These early stages of wall synthesis, detected after 18 hours, are unaffected by the presence, in the culture medium, of protein synthesis inhibitors, suggesting that synthesis of new RNA or protein is not required for wall initiation and that residual protein and endogenous hormone levels are sufficient. Structurally, the first formed envelope is amorphous and consists of pectins, but after a few days a second inner layer of cellulose fibrils is progressively laid down on the protoplast surface, eventually producing a near normal cellulose matrix after four or five days (Burgess & Fleming, 1974).
Nuclear division and cytokinesis is concomitant with cell wall formation. Occasionally cytokinesis does not occur, perhaps due to the presence of an incomplete cell wall, and gives rise to binucleate cells which may not be capable of further division.
Most protoplasts, like suspension cultured cells, have an optimum plating density as regards their division potential. At this density (usually 5 × 104 – 1 × 105 protoplasts/ml –1 ), the plating efficiency, defined as a percentage of the original plated protoplasts that produce cell colonies after 28 days culture, can reach 70%. As division proceeds, the plasmolyticum level in the culture medium is reduced stepwise by the transfer of agar blocks, containing the dividing protoplasts, to the surface of a similar medium of lower osmotic pressure. Individual cell colonies are pricked out, 4–6 weeks after plating, and subcultured onto a regeneration medium. Callus masses thus produced may undergo differentiation, via organogenesis, eventually producing plantlets. Regenerating protoplasts of certain species, such as carrot, will form embryoids in liquid culture.
Whole plant regeneration from protoplasts can be achieved for an expanding list of species of distinct taxonomic groups including the Solanaceae (tobacco, Petunia ) (Frearson et al., 1973), Brassicaeeae (rape) (Kartha et al., 1974),
Papilionaceae (pea, cowpea) and Scrophulariaceae (Antirrhinum ). Whole plant regeneration is not restricted to monocotyledons or dicotyledons, haploid or diploid plants, or to protoplasts obtained directly from whole plant tissues. The majority of plants regenerated from protoplasts are normal and exhibit a high degree of fertility, but a small and variable (less than 0.1%) number of plants show morphological abnormalities owing to aneuploidy and polyploidy.
An example of this can be seen following the regeneration of plants from mesophyll protoplasts of diploid Petunia, homozygous for blue flower colour. Some regenerated plants have a tetraploid chromosome number, which is also accompanied by a flower colour change from blue to red. Since well established protoplast cultures have high plating efficiencies, and hence efficient cloning potentials, abnormal plants can arise from the expression of cell aberrations present in the tissue prior to protoplast isolation.
The nutritional and hormonal requirements of cultured protoplasts are constantly varying depending upon the stage in the regeneration process. The photosynthetic capacity and respiration rate of the protoplast is suppressed by the plasmolysing conditions. Changes in metabolic requirements during the early stages of growth could be attributed to enzyme uptake during isolation and the rigours of cell wall synthesis. These subtle changes are highlighted in the crown gall (tumour) (Scowcroft et al., 1973), where protoplasts require an exogenous growth regulator supply for the initiation of division, unlike the tumour cells which are autotrophic for auxin. Following cell wall regeneration and division, the requirement for exogenous growth regulator supply is lost, suggesting that growth regulator autonomy is, at least in part, dependent upon the presence of a functional cell wall.
16.3—
Protoplasts and Auxin Responses
Although protoplasts require both auxins and cytokinins for the initiation and maintenance of division following cell wall formation, the freshly isolated protoplast has facilitated studies of the direct and rapid physiological action of auxins upon the plasmalemma. Auxins such as IAA are known to influence cell elongation in the plant. Leaf or coleoptile protoplasts, incubated in the presence of physiological levels of IAA, respond by an increased permeability of the plasmalemma, probably to water. Protoplasts maintained in an isotonic plasmolyticum containing auxin, undergo a rapid expansion as the internal volume increases owing to extensive vacuolation, which often results in bursting (Hall & Cocking, 1974). The presence of anti-auxins, which preferentially bind to the site of action of the auxin, suppresses the bursting response indicating that these effects are the result of auxin action. As may be expected, protoplasts isolated from tumour cells which have high endogenous levels of auxins, require significantly higher levels of exogenously supplied auxin to elicit the bursting response. Vacuoles produced by disruption of protoplasts show no response to
auxins, indicating clear differences in structure and function of the tonoplast. It is difficult to relate these responses to the auxin control of growth in the plant, but the induction of membrane convolutions, seen prior to bursting, can influence the thickness of the newly synthesized wall and hence cell shape and direction of growth.
Herbicides, such as 2,4-D, are used as growth regulators at low concentrations in culture media. There appears therefore to be a balance, for some species, between controlled growth and a herbicidal effect. Protoplasts, when incubated in the presence of herbicides such as paraquat, do not respond by expansion, but collapse as a result of disruption of the plasmalemma. It may be therefore, that control of the selective action of some herbicides is found in the plasmalemma itself.
16.4—
Uptake Properties of Protoplasts
16.4.1—
Macromolecules
During isolation, and for several hours after release, protoplasts exhibit extensive pinocytic activity. This process, which may be stimulated by the plasmolysing conditions, enables macromolecules, such as ferritin and polystyrene latex particles, to enter the protoplast. Infoldings of the plasmalemma result in the formation of vesicles which contain small quantities of the surrounding medium together with any particles that may be suspended in the medium and which are less than I µm in size. Particles entering protoplasts in this way are often accumulated in the central vacuole. These basic studies have led on to an examination of uptake in relation to cell and ultimately whole plant modification, by the incorporation into protoplasts of biologically active particles. Certain microorganisms can be transformed by the incorporation into the genome of exogenously supplied DNA, which in turn can be expressed, thus modifying the cell. Comparable attempts at transforming eukaryotic cells, involving plant protoplasts, are in progress and it is clear that DNA will enter protoplasts but no evidence for the persistent expression of this input DNA currently exists at the protoplast/cell level.
16.4.2—
Chloroplasts, Nuclei and Bacteria
A further extension of the uptake phenomenon has been successfully applied to whole bacteria and plant organelles such as nuclei and chloroplasts (Bonnett & Eriksson, 1974). The precise mechanism of entry of these relatively large particles may be one of membrane fusion, in the case of nuclei and chloroplasts, rather than pinocytosis. Conditions favouring organelle uptake are very similar to those that bring about protoplast fusion. Nitrogen fixing bacteria (Rhizobium )
can enter pea leaf protoplasts during the isolation procedure (Davey & Cocking, 1972) (Fig. 16.2e) but evidence for nitrogen fixing ability of the bacterium/plant association is still awaited. Likewise transplanted nuclei may survive degradation, and following fusion with the nucleus of the recipient cell, can result in the formation of hybrid or modified cell lines. Organelles entering protoplasts as a result of uptake into vesicles may not be able to function in a strictly symbiotic manner since they are not in direct contact with the cytoplasm. However, these novel approaches to cell modification may have major implications for the plant breeder and could possibly lead to the production of more efficient economically important plant species.
16.4.3—
Virus Particles
The availability of protoplasts in large quantities has overcome many biological problems facing the investigator of host-virus relationships. In intact plants the process of infection is not synchronous and its establishment is restricted to cells with broken or damaged cell walls (Zaitlin & Beachy, 1974). Protoplasts become infected following a short incubation with intact virus or its RNA and in the presence of a polycation, such as poly-l-ornithine, which not only damages the plasmalemma but neutralizes the negative charge common to many virus particles. This allows adsorption of particles, to the plasmalemma surface, which can then enter the protoplast through damaged regions of the membrane or via pinocytic vesicles. The process of infection varies considerably depending upon the virus, but in general, newly synthesized material, possibly RNA, is detected after 2–3 days in vesicles closely associated with the endoplasmic reticulum and nucleus. Newly formed virus particles are also found within the nuclear membrane. However, it is impossible to eliminate influences of the plasmolyticum, enzyme carryover and the absence of a cell wall upon the infection process, which may therefore be dissimilar to that in whole plants. Some protoplasts resist infection with virus, and it may be possible to regenerate plants, as described previously, from such protoplasts, which are not susceptible to virus infection.
16.5—
Somatic Hybridization of Plants
Crop improvement advances with the production of new hybrids, which, in turn, are selected for the expression of desired characteristics. However, in order that plant species can preserve their genotypes there exists major incompatibility barriers. The fusion of plant protoplasts of different species offers one approach that could by-pass the sexual cycle and, provided that nuclear fusion occurs in the resultant heterokaryon, new somatic hybrids may arise following regeneration.
16.5.1—
Induced Fusion of Protoplasts
Several methods now exist for the fusion of protoplasts and all embrace the same basic concepts. It is first necessary to bring the protoplasts together so that the two plasmalemma surfaces are in contact, whilst at the same time inducing destabilized regions on the membrane. Where two such regions of adjacent protoplasts coincide, localized membrane fusion occurs, together with limited cytoplasm mixing, to produce cytoplasmic bridges. Further mixing of the cytoplasms (Fig. 16.2c) eventually results in the complete coalescence of the protoplasts which are then returned to stabilizing conditions after removal of the fusion inducing agent.
A slightly hypotonic aqueous sodium nitrate solution reduces the negative charge on the outer membrane thus allowing adhesion of protoplasts. The presence of the Na+ ion induces localized realignment of the lipo-protein in the membrane which in turn allows fusion to occur when two such regions come into contact. Slightly deplasmolysing conditions facilitate cytoplasmic coalescence (Power & Cocking, 1971).
PEG solutions induce tight adhesion of protoplasts (Kao et al., 1974) owing to electrostatic forces. The presence of cations, such as Ca2+ , in the PEG solution, enhances adhesion since the calcium ion may form a molecular bridge between the negatively polarized PEG molecule and the protein of the membrane. Water is also withdrawn from the protoplast by PEG, thus reducing the turgidity of the system and allowing closer packing of protoplasts. This dramatic disruption, and possible reversal, of the charge properties of the membrane after removal of PEG, leads to membrane fusion.
Incubation of protoplasts in solutions buffered at high pH (Keller & Melchers, 1973) and containing calcium ions also induces fusion. This may resemble the action of PEG in reversing membrane charge properties in conjunction with the establishment of molecular bridges prior to membrane fusion.
Protoplasts isolated from pollen mother cells will fuse in the absence of an inducer. In such cases the membrane structure may be quite different to the plasmalemma of somatic cells since the products of pollen mother cell development, germinating pollen grains, are ultimately involved in the fertilization process and hence membrane fusion.
16.5.2—
Culture of Fusion Products
Following fusion treatment, protoplasts are returned to the culture medium and cell wall formation proceeds. It is during the first mitotic division that nuclear fusion can occur as a result of common spindle formation, giving rise to the hybrid cell. Abortion of the nuclei, unidirectional chromosome elimination, or the inability to produce a nuclear hybrid may be expected between species that are not closely related genetically. Following regeneration into whole plants, the resulting cytoplasmic hybrids ('cybrids') may be of immense value to the
plant breeder since many plastid characteristics and resistance to certain diseases are controlled by plasmagenes.
The frequency of true hybrid cell formation is low (1 in 105 ). This fact has led to the development of generally applicable cultural procedures aimed at preferentially selecting out hybrid cells. These techniques, currently receiving most attention by protoplast workers, are based upon an ability of the nuclei to complement in the hybrid cell. This involves the use of parental species that exhibit mutually exclusive biochemical features such as differential drug sensitivity (Cocking et al., 1974), light sensitivity or auxotrophic requirements.
Following selection, somatic hybrid plants have been produced between two Nicotiana species. In this particular case, the selection theory was based upon prior knowledge of the cultural requirements of the sexually produced hybrid (Carlson et al., 1972). Protoplasts of the parents, N. glauca, N. langsdorffi, did not grow in a culture medium that supported the growth of leaf protoplasts of the sexual hybrid. Following fusion with sodium nitrate, the protoplasts were plated in this medium and a few colonies were recovered which later developed into plants. Electrophoretic and karyotypic analysis confirmed that these plants were amphidiploid somatic hybrids.
In spite of this unambiguous proof that somatic hybridization is possible, it will only be when somatic hybridization is demonstrated for plant species which are truly sexually incompatible, that its potential for crop improvement will be fully realized.
Further Reading
Bajaj Y.P.S. (1974) Potentials of protoplast culture work in agriculture. Euphytica 23, 633–49. Cocking E.C. (1972) Plant cell protoplasts—isolation and development. Ann. Rev. Plant Physiol.23, 29–50.
Cocking E.C. (1974) The isolation of plant protoplasts. Methods in Enzymology (eds S. Fleischer & L. Packer), Vol. XXXI part A pp. 578–83.
Heyn R.F., Rörsch A. & Schilperoort R.A. (1974) Prospects in genetic engineering of plants. Quart. Revs. Biophys. 7, 35–73.
Kruse P.F. & Patterson M.K. (Eds.) (1973) Tissue Culture—Methods and Applications. Academic Press, London.
Nickell L.G. & Heinz D.J. (1974) Potential of cell and tissue culture techniques as aids in economic plant improvement. Genes, Enzymes and Populations (eds. A.M. Srb), pp. 109–28. Plenum Publishing Corp., New York.
Smith H.H. (1974) Model systems for somatic cell plant genetics. Bioscience24 (5), 269–76.
Street H.E. (Ed.) (1977) Plant Tissue and Cell Culture, second edition. Botanical Monographs Vol. 11. Blackwell Scientific Publications, Oxford.
Tempé J. (Ed.) (1973) Protoplastes et fusion de cellules somatiques végétales. Colloques Int. C.N.R.S., No. 212. Paris: Editions I.N.R.A.
Chapter 17—
Genetic Variation in Cultured Plant Cells
17.1—
Introduction
Genetic variability can be described as the quantity of alleles which exist among the total genetic loci of a population. In many crop plants genetic variability is rapidly diminishing (Day, 1972) as a result of increasingly specialized breeding, intensive agriculture, and wide dissemination of the most desirable cultivars. Serious consequences are uniform susceptibility to new pathogens, inability to adapt to new environments as well as the inadvertent spread of 'silent' deleterious genes. The southern corn blight, which destroyed one billion dollars worth of the U.S. corn crop in 1970, occurred in large part because 70% of the cultivated corn contained the same cytoplasmic determinant for male sterility. A pleiotropic effect of this gene is blight susceptibility.
While efforts are underway to preserve existing variability (e.g. in germ plasm collections) other approaches such as mutation breeding seek to increase variability. To this end plant cell culture is potentially well-suited. The plant cell geneticist can easily select certain mutants from among 104 –105 cells per ml of suspension culture. Each cell is a potential plant which, if field tested by conventional methods, would require huge investments of time, labour and acreage. The individual cell has limited metabolic pools and can be cultured on a completely defined and relatively simple medium (see chapter 15). The plant cell geneticist thus can establish a wide range of conditions to select for specific biochemical mutants which are perhaps not so easily selected in the field. The ability to select defined genetic alterations will provide a means for the dissection of biochemical and developmental pathways and for the analysis of mechanisms of genetic expression in higher plants. This knowledge will lead to efficient selection in vitro of mutations which result in improved nitrate utilization, higher heavy metal tolerance, over production of a specific amino acid or seed protein, and other valuable traits (Chaleff & Carlson, 1974). Theoretically, the use of tissue cultures from anther-derived haploid plants (see chapter 15) would allow the direct selection of recessive mutations. Finally, whole plant regeneration from callus is becoming possible in an ever-increasing number of species. This is essential if the microbial geneticist's approach in plant cell culture is to complement conventional plant breeding.
The following section discusses some mutants already obtained in culture and some advances in plant cell genetics that are important for producing additional agriculturally useful variation.
17.2—
Induced Variation and Selection
The spectrum of mutants recovered among cultured plant cells has so far been determined primarily by the growth characteristics and requirements of these cells in vitro. The strict prerequisite of a high minimum density for proliferation of cells (Street, 1973) or protoplasts (Nagata & Takebe, 1971) in vitro makes easier the selection for mutants resistant to antibiotics and antimetabolites than the selection for auxotrophs. The isolation of auxotrophs requires an experimental system composed of haploid cells in which the wild type cells are at a disadvantage compared to the mutants. This approach was used by Carlson (1970) to isolate six auxotrophic mutants of Nicotiana tabacum. The selective system was adapted from a procedure used successfully with mammalian cells (Puck & Kao, 1967). In a minimal medium containing 5-bromodeoxyuridine (BUdR), actively dividing prototrophic cells will incorporate more of this lightsensitive nucleoside analogue into DNA than will arrested auxotrophs. The auxotrophs will survive subsequent illumination at a much higher frequency than the prototrophs and will form clones on a supplemented medium. The mutant tobacco calluses which were isolated grew slowly on a minimal medium. Plants were differentiated from four of the mutant clones and these also grew slowly without supplementation. In genetic analyses of these four regenerated plants, three plants transmitted the mutant phenotype as a single recessive Mendelian factor and one displayed a more complex pattern of inheritance. The recovery of only leaky mutants may be due to a lack of functional diploidization of the N. tabacum genome. Haploid cells obtained from this amphiploid species may contain two copies of many metabolically essential genes and thus two mutational events may be required to delete completely certain functions (Carlson, 1970).
Cell lines which exhibit increased resistance to a wide variety of antimetabolites have been isolated and studied. Haploid cultures of petunia (Binding et al., 1970; Binding, 1972) and tobacco (Maliga et al., 1973) resistant to streptomycin have been selected. In genetic crosses involving a diploid plant obtained from one mutant tobacco clone, drug resistance segregated as a maternally inherited character (Maliga et al., 1973; Sz.-Breznovits et al., 1974).
Plant cell lines containing dominant or semidominant genetic markers may prove useful in selecting hybrid cells formed by protoplast fusion. This realization has motivated the isolation of cell lines resistant to 8-azaguanine and BUdR since such mutants are employed in selecting hybrids in mammalian cell systems. Resistance to these analogue precursors of nucleic acids in cultured mammalian cells may result from deficiencies in hypoxanthine:guanosine phosphoribosyl transferase (HGPRT) and thymidine kinase activities, respectively, or in the uptake of these analogues (Kit et al., 1963; Littlefield, 1964a; Breslow & Goldsby, 1969). In the presence of aminopterin, endogenous biosynthesis of thymidine and hypoxanthine is inhibited and cultured mammalian cells are dependent upon an exogenous supply of these substances (Fig. 17.1). As each
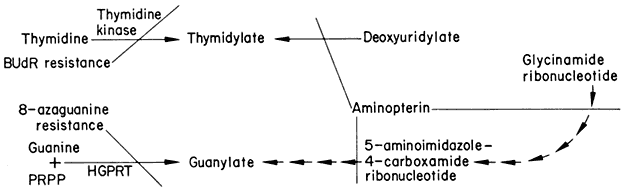
Figure 17.1
Simplified scheme showing pathways for synthesis of guanylate and thymidylate.
Endogenous biosynthetic steps blocked by aminopterin-inhibition of tetrahydrofolate
synthesis are shown as well as the genetic blocks which confer resistance to BUdR and 8-azaguanine.
mutant cell line is unable to utilize one of these compounds, only complementing hybrids formed by cell fusion survive in a medium containing hypoxanthine, aminopterin, and thymidine (the HAT system) (Littlefield, 1964b). Although a low concentration of aminopterin inhibits growth of sycamore cells (Bright & Northcote, 1974) it should not necessarily be assumed that this effect is caused by the same mechanism in both animal and plant systems. The isolation of cell lines of tobacco (Lescure, 1973) and sycamore (Bright & Northcote, 1975) resistant to 8-azaguanine has been reported. The resistance phenotype is not completely stable in the sycamore cell line in which HGPRT activi ty is half that of the wild type level. The latter result is not unexpected in a diploid system and emphasizes the necessity of using haploid cell lines in order to recover mutants with an anticipated recessive phenotype.
Resistance to BUdR has been selected in haploid tobacco (Maliga et al., 1973) and in presumably diploid sycamore (Bright & Northcote, 1974) and soybean (Ohyama, 1974) cells. The resistant sycamore and soybean cultures did not differ significantly from the wild type in uptake of thymidine or in the level of thymidine kinase activity (Bright & Northcote, 1974; Ohyama, 1974) and the basis for the resistance phenotype remains unknown. Marton and Maliga (1974) have observed transmission of BUdR resistance to progeny of plants derived from one mutant tobacco callus and unimpaired uptake and incorporation of thymidine in four resistant cell lines.
Heimer and Filner (1970) have isolated a cell line of tobacco which is insensitive to inhibition by threonine when grown in a medium containing nitrate as sole nitrogen source. By studying nitrate accumulation in this mutant under conditions in which nitrate reductase is chemically inactivated and in which nitrate uptake by wild type cells is repressed, it was concluded that regulation of nitrate uptake is altered in the resistant cell line. Such mutants should prove extremely useful in the investigation of nitrogen metabolism.
The potential agronomic importance of tissue culture techniques is illustrated by experiments in which screening of cultured somatic cells resulted in the recovery of mutants of N. tabacum resistant to wildfire disease (Carlson,
1973a). This disease of tobacco is caused by a bacterial pathogen, Pseudomonas tabaci, which produces a toxin structurally related to methionine (Stewart, 1971; Fig. 17.2). Populations of mutagenized haploid cells were plated in a medium containing an inhibitory concentration of methionine sulphoximine, an analogue of the wildfire toxin (Braun, 1955) and resistant clones were selected. Three homozygous diploid plants which were regenerated from these methionine sulphoximine-resistant calluses are less susceptible than the parent plant to the pathogenic effects of bacterial infection. One mutant plant contains wild type intracellular levels of free methionine and resistance appears to be genetically complex. In the other two resistant plants the endogenous concentration of free methionine is five times higher than in the wild type while the free pool sizes of other amino acids remain unchanged (Carlson, 1973a). The levels of free methionine in the heterozygous plants are intermediate between those in the wild type and in the homozygous mutant (Chaleff & Carlson, 1975a).
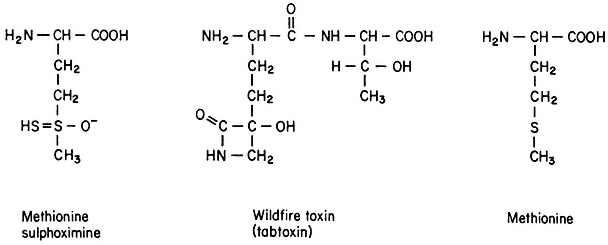
Figure 17.2
Structures of methionine sulphoximine,
wildfire toxin (Stewart, 1971), and methionine.
Intermediate methionine levels are consistent with the observed transmission of the resistance phenotype of these two plants as a single semidominant locus (Carlson, 1973a). These experiments suggest that selection for toxin resistance in vitro may provide a generalized procedure for generating disease resistant varieties. It is also evident that regulation of amino acid metabolism may be altered in cells selected for resistance to an amino acid analogue and that this mutational event may be expressed stably in mature plants derived from the mutant callus tissue. Additional support for this conclusion is provided by other research.
Widholm (1972a,b) has isolated presumably diploid cell lines of N. tabacum and Daucus carota which are capable of growth in the presence of a normally inhibitory concentration of 5-methyltryptophan. Crude extracts of the resistant cell lines (5-mtr ) contain a species of anthranilate synthetase which is less sensitive to feedback inhibition by tryptophan and 5-methyltryptophan than is the wild type enzyme. Endogenous levels of free tryptophan in 5-mtr cell lines of tobacco and carrot are 15 times and 27 times higher, respectively, than the wild
type levels. Plants could not be regenerated from the tobacco and carrot cells which synthesized a feedback-insensitive form of anthranilate synthetase. However, plants were obtained from analogue-resistant carrot cultures in which anthranilate synthetase activity was indistinguishable from the wild type and uptake of 5-methyltryptophan and tryptophan was reduced. Although no genetic studies with these plants have been reported, cell lines re-initiated from the plants maintained the same degree of resistance to 5-methyltryptophan as the original selected lines. Cell cultures of tobacco and carrot resistant to p -fluorophenylalanine were isolated also. Resistance to this analogue apparently resulted from diminished rates of transport and incorporation (Widholm, 1974).
Two attempts have been made to employ analogue resistance as a selective screen in obtaining callus cultures of cereals which overproduce amino acids essential to human nutrition. Chaleff and Carlson (1975b) recovered three cell lines of rice which were stably resistant to the lysine analogue S-2-aminoethylcysteine. Analyses of both free pool and total amino acid compositions showed that the resistant cultures contained elevated levels of several amino acids, including lysine, methionine, leucine and isoleucine. Cheng (1975) has selected cultures of Hordeum vulgare which are resistant to the same analogue and in which lysine levels were increased, as was the rate of incorporation of exogenously supplied lysine. Unfortunately, attempts to differentiate plantlets from the variant calluses were unsuccessful in both cases. Although much valuable information is to be obtained from research with variant cell lines, such studies are severely limited by the inability to examine genetic expression and transmission in the mature plant. Future investigations should attempt to utilize experimental systems in which plants may be differentiated from the altered cell lines.
17.3—
Introduced Variation—Gene Transfer
17.3.1—
Introduction
The systems so far described have dealt with mutationally-induced variation of the pre-existing plant genome. Considering such factors as genome size, multiple gene copies (e.g. non-allelic isozymes), and the tendency of plants to become aneuploid and polyploid it should be apparent that the potential for induced variation in plant cells is enormous. As culturing and selective techniques improve and a better understanding of plant biochemistry is attained (largely via tissue culture) the spectrum of plant cell mutants recovered should widen.
In addition, manipulation of plant cells offers unique opportunities to introduce foreign genetic information not only from other plants but also from bacteria and bacterial viruses and other organisms. In nature, non-sexual gene transfer between plants by way of DNA transformation or viral-mediated transduction has not been demonstrated. Neither has gene transfer between bacteria
and plants been demonstrated in nature although legume-Rhizobium N2 fixing symbioses and bacterial-induced crown gall tumours may disprove this contention when more is learned about these systems. However, during the last 5 years several experimental demonstrations of gene transfer in cultured plant cells have been at least partially successful, thus holding the promise that introduced as well as induced variation may become a tool in plant breeding.
Doy, Rolfe and Gresshoff (1973a) used the term transgenosis to describe the asexual introduction of foreign genes into plant cells. They emphasize that such genes could be introduced using isolated nucleic acid or an intact virus, organelle, cell or protoplast. While they include whole plants and both animal and plant cells as potential recipients the treatment here is restricted to the transgenosis of cultured plant cells and protoplasts. The use of protoplasts as simultaneous donors and recipients (protoplast fusion) has been discussed in the previous chapter.
17.3.2—
Isolated DNA-Mediated Transgenosis
Theoretically, isolated plant DNA is a logical choice as donor in transgenosis of plant cells. It has a better probability than bacterial or viral DNA of exhibiting homology with specific stretches of the host chromosome thus maximizing pairing and integration. The transcriptional and translational machinery of the host is probably better suited for plant DNA than for bacterial or phage DNA. However, the following transgenosis experiments point out the many technical difficulties in using isolated DNA in general and plant DNA in particular. Both plant and bacterial DNA are highly susceptible to plant cell nucleases. A plant DNA preparation in addition is much more dilute for a given gene than a bacterial preparation. Further, transgenosis by isolated plant DNA is currently hampered by a lack of suitable, biochemically characterized selective markers. Early experiments demonstrated a poor efficiency of DNA uptake by cultured cells and protoplasts. Bendich and Filner (1971) found that only 0.5% of isotopically labelled tobacco DNA or heterologous Pseudomonas aeruginosa bacterial DNA was taken up into the nuclear fraction of cultured tobacco cells. The rest was rapidly degraded in spite of extensive washing and pronase treatment to reduce extra-cellular DNase activity. No evidence was obtained for the integration of this DNA into the recipient genome.
Ohyama et al., (1973) suggested that protoplasts, which lack cell walls, may be better recipients of isolated DNA. In their experiments approximately 1% of exogenous E. coli DNA was found in an acid precipitable fraction of protoplasts of soybean, carrot and Ammi visnaga. By treating tobacco cells with cationic DEAE dextran, Heyn and Schilperoort (1973) increased 'uptake' of acid precipitable Agrobacterium tumefasciens DNA to 10–15% of input. Possibly DEAE Dextran and anionic DNA form a DNase resistant complex. However, only 0.145% of this DNA penetrated the cell; the rest was adsorbed to the cell wall.
Studies involving the selection of protoplasts and cultured cells which express a donor DNA marker have so far been few and only marginally successful. Holl et al., (1974) recently described two experimental systems. One involves the acquisition of mannitol utilization by soybean protoplasts from the bacterium Azotobacter vinelandii. No clear-cut DNA-mediated transgenote has yet been reported. A serious difficulty in this experimental system is the impermeability of soybean protoplasts to mannitol; thus, transgenotes would have had to acquire at least two bacterial genes—a mannitol permease as well as the gene responsible for the mannitol-fructose conversion. Such double transgenotes would probably be exceedingly rare; moreover it is difficult to envisage a bacterial permease functioning in a plant cell membrane.
In the second system described by Holl and collaborators, seeds of a mutant pea strain unable to form root nodules with Rhizobium bacteria were treated with DNA extracted from a strain which forms functional nodules. They observed that 1–2% of those plants grown from treated seeds were corrected for nitrogen-fixation. An analysis of this result should consider the mutagenic effects of DNA and the observation that all corrected seeds simultaneously recovered two lost functions which are encoded by independently segregating genes viz. nodulation and N2 -fixation. A serious restriction of this system is the limited number of seeds which can be treated. Alternatively, the authors have proposed treating cultured mutant shoot tips with DNA from the symbiosis-competent line. Transgenotes can be selected by whole plant regeneration from shoot tips on an N-free medium inoculated with Rhizobium. However, the cultured shoot tip consists of a heterogeneous population of partially differentiated diploid cells and it is not known if only certain types or minimal numbers of these cells must be transgenosized before the regenerated plant will fix nitrogen. As yet no transgenotes have been reported.
Ferrari, working in Widholm's laboratory (1975), has used carrot cell lines resistant (5mtr ) and sensitive (5mts ) to 5-methyl tryptophan to study DNA transgenosis. The transfer of genetic information was monitored by observing the frequency of 5mtr clones arising among a population of 5mts cells which had been treated with DNA extracted from 5mtr cells. Although an apparently mutagenic effect of both 5mt r and 5mts DNA contributed to altering the phenotype of the recipient cell, intact 5mtr DNA was 2 to 8 times more effective than 5mts DNA in conferring resistance to 5-methyl tryptophan. However, the observation that these differences occurred in only one quarter of the experiments illustrates the difficulties of this system. An understanding of the means by which input 5mtr DNA. effects 5-methyl tryptophan resistance awaits a biochemical comparison between donor and acquired gene products.
Obviously refinements are needed in future DNA transgenosis systems. These would include the transfer of single gene, biochemically characterized, markers to recipient cells incapable of mutational escape from selective conditions, the means of neutralizing recipient DNAases or otherwise improving recipient cell competence, and enrichment of donor (especially plant) DNA for
specific genes (e.g. by fractionation, use of organelle DNA, molecular cloning or reverse transcriptase products of purified plant messengers). Further, if isolated protoplasts are best suited for DNA uptake, callus and whole plant regeneration must be achieved from protoplasts of the many species where this is not yet possible.
17.3.3—
Viral-Mediated Transgenosis
In viral-mediated transgenosis foreign genetic information is transferred to and expressed by plant cells following exposure to intact viral particles. The experiments described here deal exclusively with bacteriophage infection although the productive infection of plant protoplasts with plant viruses is, in a strict sense, also viral mediated transgenosis. In a series of experiments Doy, Gresshoff and Rolfe (1973b) took advantage of the inability of tomato callus to survive with either lactose or galactose as the sole carbon source. In one experiment, high titre preparations of the specialized transducing bacteriophages ø80 and l were applied to tomato callus. The former (ø80 plac+ ) carried the bacterial gene coding for b -galactosidase, the enzyme which catalyses hydrolysis of lactose to glucose and galactose. The latter (l pgal+ ) carried the bacterial structural genes encoding the enzymes which catalyse the conversion of galactose to utilizable glucose 1–P. Eighty per cent of the calluses treated simultaneously with both phages or with ø80 plac+ alone were able to survive and grow slowly on lactose medium as compared with 5–10% of untreated controls.
In another experiment galactose conversion was shown to be dependent on intact bacterial gal genes since bacteriophage with no gal genes (ø80) or with a mutated gene for galactose utilization (lpgal– ) did not elicit growth with galactose as sole carbon source in the inoculated calluses.
More convincing molecular proof of bacterial gene expression by the treated plant cells was the immunological identification of the b -galactosidase activity in the lactose utilizing callus. Antiserum specific for the E. coli enzyme afforded the same degree of protection against thermal inactivation of the b -galactosidase activity in extracts of both E. coli and the lactose utilizing callus (Table 17.1). Doy, Gresshoff and Rolfe (1973a,b) have emphasized that the definition of transgenosis implies neither a specific mechanism of viral transduction of bacterial genes nor the integration and stable transmission of the acquired genes.
Carlson (1973b) demonstrated the appearance of two early phage enzymes upon 'infection' of barley protoplasts with the lytic bacteriophage T3. The phage exposed protoplasts synthesized S-adenosyl methionine cleaving enzyme (SAMase) and RNA polymerase, two T3-encoded enzymes not normally produced by the plant. The latter enzyme was definitely assigned a bacteriophage origin since exposure to T3 containing an amber (nonsense) mutation in the RNA polymerase structural gene resulted in significant protoplast synthesis of SAMase activity only. It is interesting that the phage-borne bacterial gal mutation introduced into tomato callus (Doy et al., 1973b) was also amber, providing
| |||||||||||||||||||||||||||||||||||||||||||||
circumstantial evidence that plants as well as bacteria may recognize the amber codon as a stop signal. Doy et al., (1973b) demonstrated that both tomato and Arabidopsis thaliana calluses die quite rapidly upon exposure to ø80 phage carrying a bacterial amber suppressor. It is, however, difficult to distinguish whether the bacterial suppressor tRNA interferes with termination of translation or simply competes with plant tRNA at sense codons.
It has been demonstrated (Johnson et al., 1973; Grierson et al., 1975) that sycamore cells in suspension can acquire the ability to utilize lactose from l phage carrying the lac genes. Ordinary l does not confer lactose utilizing ability. However, biochemical evidence for the presence of bacterial enzyme in cells growing on lactose has not yet been obtained. The lac+ sycamore cell lines as well as the lactose-utilizing tomato callus lost their new function in succeeding generations (Doy, Gresshoff & Rolfe, 1973b; Grierson et al., 1975). Polacco et al., (1974) demonstrated the expression of l borne lac genes in tobacco diploid callus both by growth and biochemical criteria. However, these transgenosized lines uniformly lost all lac functions after several months. These results demonstrate the transient nature of bacterial and viral gene expression in plant cells.
We may tentatively summarize the advantages and drawbacks of viralmediated transgenosis. The genome size of a transducing phage is small (l = 31 × 106 daltons) so that the desired bacterial gene is enriched about 80 fold compared with its concentration in the E. coli genome[*] . Therefore, its chances of uptake and expression are improved compared with isolated E. coli DNA transgenosis. Further, the phage coat may serve to protect the foreign DNA from nuclease attack and even provide some transport function. On the negative side the bacterial genes introduced probably lack homology with the plant genome thereby minimizing the probability of chromosomal integration and stable transmission. Of course it cannot be assumed that the transcription and translation of the bacterial genes by the plant host are efficient. Presumably, many phage specific genes are expressed in the recipient plant cell and this may be related to the generally deleterious effect of phage treatment on callus growth. The systems employed were far from ideal in that T3 genes conferred no selective advantage on barley protoplasts while calluses of tomato (Doy et al., 1973b; Table 17.1) and tobacco (Polacco et al., 1974; Fig. 17.3) and sycamore cell suspensions (Grierson et al., 1975) have been shown to have plant-specific b -galactosidase activity. As a result the selective pressures in the lplac system were probably not great; Doy et al., (1973b) reported that 5–10% of control calluses grew on lactose. The presence of a homologous plant enzyme also complicates the identification of the introduced gene.
Figure 17.3
The presence of plant specific b -galactosidase activity in extracts of tobacco
callus grown on sucrose (
to lactose (

(From Polacco et al., 1974.)
Further experiments should utilize biochemically characterized markers which confer a more rigorous selective advantage. Whole plant regeneration should be attempted under selective conditions and modes of gametic transmission of the desired trait studied. Ideally, to improve homology (and therefore integration), plant rather than bacterial genes should be introduced. In viralmediated transgenosis the problem is to incorporate specific plant DNA stretches in a phage coat. A potential system would be the incorporation of chloroplast DNA in l phage by techniques recently described (Thomas et al., 1974); such phage could possibly be used to transgenosize albino callus to green.
By involved biochemical and genetic manipulation Thomas et al., prepared l DNA (lgt ) with a 27% deletion (Fig. 17.4). It has lost the ability to lysogenize E. coli but can still replicate, produce coat protein and lyse its host. However, because the lgt DNA is smaller than normal it cannot associate with the coat protein in cell lysates to form an infective particle. The l gt DNA, therefore, cannot form plaques in transfection experiments, unless DNA from apparently any source, is biochemically inserted into the genome. Such DNA is replicated along with l DNA. Cloning such DNA-rescued phage is equivalent, then, to cloning a small stretch of the foreign DNA incorporated in the l genome. The legend to Fig. 17.4 provides more experimental detail.
Figure 17.4
Schematic procedure for incorporating small stretches of DNA from any
source (e.g. plants) in phage l (Thomas et al. 1974.) The procedure takes advantage
of the properties of the restriction endonuclease enzyme EcoR1. This enzyme recognizes
very specific nucleotide sequences (the six in l are labelled R1-1, R1-2, etc.) and cleaves them
to yield cohesive ends (short single stranded homologous regions) which can be annealed and
rejoined in vitro. EcoRi is produced by an E. coli R factor; such an E. coli is a restrictive host for l
with unmodified Rl sites. This is the basis for the selection of mutation in sites R1-4 and R1-5 in step (ii) above.
17.3.4—
Conclusion
This chapter has reviewed recent advances in the development of techniques for modifying the genetic information of cultured cells of higher plants. We have not considered nuclear and organelle uptake by protoplasts or protoplast fusion as means of introducing genetic variation (Chaleff & Carlson, 1974). The selective systems would be similar to at least some of those already described and the techniques involved are discussed in chapter 16. It is hoped that all these experimental approaches will prove useful in the elucidation of the biological processes of higher plants. These procedures also may contribute to the development of improved crop varieties. However, successful realization of these aims will require a fuller development of the systems and methods which have been described. The differentiation and chromosome composition of cultured cells must be better controlled and the minimum cell density required for growth must be reduced. In addition, these in vitro techniques must be extended to a greater number of crop species.
We should not close this chapter without mention of the potential biohazards of gene transfer experiments. The experimenter should consider potential dangers to agriculture, the ecosystem and human beings. We shall draw one possible scenario: a researcher succeeds in recovering intact infective bacteriophage from viral transgenosized callus. Subsequently the virus is found able to infect whole plants and cause severe disease symptoms and necrosis—the latter due possibly to a bacterial toxin gene or a plant cellulase gene, etc. The authors believe, however, that with proper precautions such experiments have the future potential of yielding new and valuable agricultural varieties.
Further Reading
Carlson P.S. (1970) Induction and isolation of auxotrophic mutants in somatic cell cultures of Nicotiana tabacum. Science168, 487–89.
Carlson P.S. (1973) Methionine sulphoximine-resistant mutants of tobacco. Science 180, 1366–68.
Chaleff R.S. & Carlson P.S. (1974) Somatic cell genetics of higher plants, Ann. Rev. Gen. 8, 267–78.
Doy C.H., Gresshoff P.M. & Rolfe B.G. (1973a) In: The Biochemistry of Gene Expression in Higher Organisms (eds J. Pollak & J.W. Lee) pp. 21–37. Aust. and New Zealand Book Artarmon.
Doy C.H., Gresshoff P.M. & Rolfe B.G. (1973b) Biological and molecular evidence for the transgenosis of genes from bacteria to plant cells. Proc. Nat. Acad. Sci. U.S.A.70, 723–26.
Johnson C.B. & Grierson D. (1974) The uptake and expression of DNA by plants. In: Current Advances in Plant Science (ed. H. Smith), vol. 2, No. 9, 1–12.
Heyn R.F., Rörsch A. & Schilperoort R.A. (1974) Prospects in genetic engineering of plants. Quarterly Review of Biophysics7, 35–73.